









Abstract
Genome sequencing of cancer and normal tissues, alongside single-cell transcriptomics, continues to produce findings that challenge the idea that cancer is a ‘genetic disease’, as posited by the somatic mutation theory (SMT). In this prevailing paradigm, tumorigenesis is caused by cancer-driving somatic mutations and clonal expansion. However, results from tumor sequencing, motivated by the genetic paradigm itself, create apparent ‘paradoxes’ that are not conducive to a pure SMT. But beyond genetic causation, the new results lend credence to old ideas from organismal biology. To resolve inconsistencies between the genetic paradigm of cancer and biological reality, we must complement deep sequencing with deep thinking: embrace formal theory and historicity of biological entities, and (re)consider non-genetic plasticity of cells and tissues. In this Essay, we discuss the concepts of cell state dynamics and tissue fields that emerge from the collective action of genes and of cells in their morphogenetic context, respectively, and how they help explain inconsistencies in the data in the context of SMT.
Citation: Huang S, Soto AM, Sonnenschein C (2025) The end of the genetic paradigm of cancer. PLoS Biol 23(3):
e3003052.
https://doi.org/10.1371/journal.pbio.3003052
Published: March 18, 2025
Copyright: © 2025 Huang et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: This work was supported by the U.S. National Institutes of Health, NIH (National Cancer Institute R01CA255536-01A1 to SH, National Institute of General Medical Sciences R01GM135396-01 to SH, National Institute of Environmental Health Sciences ES030045 to AMS) and by the Cancer Research UK (Cancer Grand Challenge, C67229/A29068 to SH). Additional support to AMS was provided by the Templeton Foundation (grant 62220, subaward A009723018), the FREIA project of the EU Horizon 2020 Research and Innovation programme (http://freiaproject.eu/wp/; no. 825100) and in-kind support by the Institute for Advanced Studies of Nantes. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have no competing financial interests to declare.
Abbreviations:
CAFs,
cancer-associated fibroblasts; ECM,
extracellular matrix; GRN,
gene regulatory network; NMU,
N-nitrosomethylurea; SMT,
somatic mutation theory; TFs,
transcription factors; TME,
tumor microenvironment
Introduction
It is said that the wise only believe in what they can see, and the fools only see what they can believe in. The latter attitude cements paradigms, and paradigms are amplified by any new-looking glass that puts one’s way of seeing the world on steroids. In cancer research, such a self-fulfilling prophecy has been fueled by next-generation DNA sequencing. The ease of access to sequencing has stimulated the unfettered quest for genetic alterations in tumor cells that would explain cancer. Since the 1970s, the elevation of the concept of ‘cancer as a genetic disease’ [1–4] has been driven by a number of factors, including discoveries of cancer-associated mutations, narrowly gene-engineered mouse models, funding policies that follow crowd thinking, and the promise of targeted therapy. The propensity of the human mind to cling to simple, mechanistically plausible explanations further promoted narratives of oncogenic ‘driver mutations’ [5–7]. The somatic mutation theory (SMT) and its tacit claim to represent the “truth” on the origin of cancer is concisely articulated in this quotation by M. Stratton and L. Alexandrov from a decade ago:
“All cancers originate from a single cell that starts to behave abnormally due to the acquired somatic mutations in its genome” (2014). [8]
This quotation dates back to 2014—and we will present an updated 2024 version later. However, during the past decade, widespread acceptance of the SMT has driven the use of next-generation sequencing. With lowering costs of genome analysis, the genetic paradigm led to ‘precision oncology’; but targeting driver mutations has invariably met the challenge of relapse of treatment-resistant cancer, too fast and in far more aggressive form [9–13] than can be explained by selection for new mutants. Knowing the mutational profile of a patient’s tumor genome has yielded little benefit in ‘personalized/precision’ cancer care [7,9,14,15].
Ironically, over the past years, the same sequencing technology has exposed new cracks in the edifice of the cancer genetics paradigm. Table 1 summarizes the most salient findings that challenge the paradigm. Some sequencing results are compatible with the postulate of oncogenic mutations, but many inconvenient findings are overlooked for lack of critical discourse, resulting in an unbalanced view. Thus, an open discussion of the growing sequencing data that are contradictory to the genetic paradigm is due. The (apparent) paradox goes both ways: many cancers harbor no consistent driver mutations, while canonical oncogenic mutations are found in tissues that remain free of cancer [16] (see Table 1 for references). Paradoxes, according to Nils Bohr, are of course central for progress [17]—if properly recognized.
Large-scale cancer genome sequencing endeavors, such as The Cancer Genome Atlas (TCGA) [82], launched in 2006 by the U.S. National Institutes of Health, have uncovered a mesmerizing breadth of genetic mutations associated with cancer. The vast, nearly chaotic diversity of genetic alterations found within the same nominal tumor type between patients is staggering and questions a deterministic logic of causation of cancer by mutations in oncogenic pathways (Table 1). However, patterns, such as recurrent mutational signatures and chromosomal rearrangements, are characteristic for distinct tumor types and stages [53,83] and point to a complex interplay between stochastic elements and some rule-governed biological causation.
The immense diversity of functions of the (mutated) molecular pathways allegedly linked to cancer causation [21], and the fact that cancer is a robust, universal phenomenon amongst metazoans, tells a story of principles of development and evolution of multi-cellular life behind the very existence and the near- inevitability of neoplasia, transcending phylogenesis, ontogenesis and oncogenesis [84–94]. However, in the quest for predictive biomarkers and molecular targets, the cancer research community has abandoned deep thinking for deep sequencing, interpreting data through the lens of clinical translation detached from fundamental biology.
The core of the genetic paradigm: Somatic Darwinian evolution
To revisit the role of mutations in carcinogenesis, we need to recall a central corollary of the cancer genetics paradigm that links somatic mutations to the cancer phenotype and cancer progression: namely, the idea of somatic Darwinian evolution of the mutated cells. Herein, random mutations may (by chance) confer functional advantages, e.g., promote autonomous proliferation that increase cell “fitness” [95]. Then, under natural selection in the cell population, those cells carrying mutations that confer a “fitter” phenotype would clonally expand, with the “fittest” clone (derived from a single cell) eventually taking over the population. This Neo-Darwinian scheme is applied to tumors in a loose, qualitative, if not figurative manner [96], often departing from rigorous concepts of evolutionary biology, and eventually becoming known as the SMT [97]. The idea of SMT has its origin in 1914 when the embryologist Theodor Boveri proposed that cancer originates from chromatin alterations in a cell. It was later recast in terms of accumulation of a number of genetic mutations by Bauer and Nordling in the 1950s [98, 99], implying a need for increased cellular proliferation that is acquired as a novel trait—but still without invoking Darwinian positive selection.
The concept of carcinogenesis as an explicit process of (somatic) evolution in the Neo-Darwinian sense was popularized by Nowell’s seminal paper in 1976 [2]. However, he introduced a pluralistic view:
“…this correlation between observable genetic change and tumor progression does not, of course, prove causality, and some workers believe that at least some aspects of the evolutionary process, (…) may represent epigenetic, rather than genetic phenomena.”
These cautionary words have been largely left out of the equation of cancer genetics in the following 50 years. Despite its rapid ascendance, SMT remains a conjecture supported by spotty, selective evidence rather than a precisely formulated theory, notwithstanding its use of mathematical formalisms [100]. Specifically, SMT left out broader considerations of historicity (ontogenesis and phylogenesis) and physiology. First, autonomous proliferation appears to be a latent ‘default’ state, ‘baked-in’ in organismal existence already in the last universal common ancestor, and is not an evolutionary novelty that is attained through natural selection in the tissue [101,102]. Second, expansion of faster proliferating cell clones is not limited to tumors, as evident in aging normal tissue and chronic diseases where hyperproliferation results in oligo-clonal expansion [103–105]. Yet, convenient for its simplicity, but misleading in its meaning, the Neo-Darwinian principle has become the unquestioned truth in tumor biology which, as Table 1 shows, is responsible for many of the cracks in the genetic paradigm of cancer.
Robust manifestation of natural selection in tumors would be ‘hard selective sweeps’ [106,107]: the complete overtaking of the tumor cell population by a malignant clone carrying the driver mutation (along with linked, but neutral mutations), representing one step in Vogelstein’s multi-step progression model of linear evolution [4,46]. The genetic fixation (as it is called in population genetics, see Table 1, point 6) of the oncogenic allele in the tumor cell population would be required for the logics of precision oncology that seeks to target a mutation to eradicate a tumor. In reality, a tumor contains thousands of genetically distinct, apparently independent cell clones, giving rise to genetic intra-tumor heterogeneity that came as a shock to the precision oncology world [37,42]. This finding led to attempts of adjusting precision oncology by targeting “trunk mutations” shared by every cancer cell and by combination therapy directed against multiple oncogenic mutations—approaches that still adhere to SMT [108].
But, even if a clone appears to expand faster and dominate the tumor, there is little proof that a complex tumor phenotype is sculpted by natural selection for specific oncogenic mutations that produce said ‘adaptive traits’ as Neo-Darwinism postulates. It is likely correct that simple Darwinian evolution is at display in the case of evasion from targeted therapy, notably kinase inhibitor drugs [41], wherein a point mutation in the kinase protein interferes with multiple aspects of kinase activity energetics, as best studied in the oncoprotein CBR-Abl, as it becomes resistant to the highly selective inhibitor imatinib [109]. But, adaptive selection of such molecular phenotype is a far cry from the suggested generation of complex traits that embody the “hallmarks of cancer”[110,111] by natural selection during tumor progression. Thus, SMT suffers from the same shortcoming as Neo-Darwinian evolution [112] in that it only explains distinct localized adaptive traits obviously linked to the selection pressure but not the emergence of complex, higher-order biological functions.
One must also not forget that neutral variants can be enriched without being selected for (Table 1, points 5, 6) because of (random) genetic drift in relatively small cell populations. In finite cell populations the mathematics of random birth-death processes alone accounts for highly skewed allele frequency distributions that defy our numerical intuition (“the rich get richer—without being more capable”) [39,113,114], although neutral drift may be overestimated because of late appearing subclonal driver mutations [115].
Complicating the picture of somatic evolution are interactions at a higher level, like those between subclones of cancer cells that affect fitness of the entire tumor [116–119], creating non-cell autonomous dynamics [104]. Beyond direct mutual stimulation, the distinct clones may functionally complement each other because of their ‘specialist’ capabilities that support the cancer cell community, for instance, through metabolism that alters pH and nutrients in the intercellular space, detoxification, promotion of angiogenesis, alteration of tissue mechanics [105], or averting anti-tumor immunity. These biological functions are not sufficient to grow a tumor themselves. Conversely, some tumor cells may keep their more malignant neighbors at bay and must not be killed [106].
Thus, if we view the tumor as an evolving ecosystem, as has become fashionable, we also must embrace evolutionary/ecological principles that have emerged in the past few decades and that question the dominance of the simple scheme of ‘natural selection’ of Neo-Darwinism [55,120–124]. These include punctuated equilibrium (stasis with sudden bursts of evolutionary events) [125,126], group selection (for fitness of a group due to social behaviors) [126,127], niche construction (evolving organism modifies its own selective environment) [128] and structuralism (natural selection must obey constraints of physics, chemistry and geometry, etc. which contribute to shaping the phenotype with no selection) [129].
Of particular relevance is the group of evolutionary theories that emphasize phenotypic plasticity [123,130,131]. These theories all suggest that the direction of evolutionary adaptation of populations ‘tracks’ the physiological adaptation of the individual to its environment as enabled by regulated phenotype plasticity, such as acclimatization. These theories include genetic assimilation/genomic accommodation [123,130–132], the Baldwin effect and canalization [133,134], and phenotype-first dynamics [94,135]. These mutation-less mechanisms may ‘lubricate’ and channel natural selection; thus, they actually support Darwinian natural selection by making evolution of adaptive traits more likely in view of the ‘unlikely’ mutations that need to happen. Thus, despite being largely absent in SMT literature, all these theories indirectly support Darwinism by considering non-genetic plasticity, thereby relaxing the rigid one-genotype-one-phenotype constraint.
Overcoming the limitations of the current paradigm: Expand it or replace it?
Calls for overcoming the blinders of the genetic paradigm have a long history. What is new is that, first, the same omics technologies employed to identify new oncogenic mutations have also been used to better understand non-genetic processes in tumorigenesis that involve cell plasticity and tissue organization. And second, the quest for drug targets or biomarkers have meanwhile reduced much of cancer research from a scholarly biological discipline to a transactional operation concerned more with cataloguing molecular alterations and finding associations, than with understanding fundamental principles using formal theory [136]. The zest for intellectual discourse on the nature of cancer, the type of scholarly discourse that advances all scientific disciplines, is long gone, or viewed upon with suspicion.
Since one should not postulate the end of a paradigm without specifying the ‘alternatives’ that may correct or replace it, we will discuss here two sets of ideas that have been sidelined by the dominance of the genetic paradigm of cancer. First, that “cancer is not a disease of the genes”, but of gene regulation and thus, of the cell. We must consider the dynamics of the collective action of genes in a network that governs cell behaviors. Second, that ‘cancer is not a disease of the cell’, but of tissues [137]; we must consider principles of tissue organization. Both of these ‘alternatives’ are discussed in detail below.
To interpret observations afforded by powerful new technologies, we more than ever need a solid framework grounded in biological principles that must be rigorously formulated (but may not yet be mathematically formalized) [138]. Cancer research needs to return from current transactional activities to the scholarly investigations enjoyed by generations of biologists who have placed new findings in the context of organisms, their development, physiology and evolution [136,139]. As Philip Ball points out, the apparent lack of ‘progress’ in the biological sciences may be related to the adoption of a mistaken paradigm [140]. Herein, we will illuminate the paths out of the genetic paradigm, touching on scientific, epistemological and sociological aspects of cancer research.
History of anomalies before the era of molecular genetics: The normalization of cancer
If so many sequencing results are inconsistent with SMT, why do they not trigger a rethink among cancer researchers? Revisiting an overused idea of the philosopher Thomas Kuhn on how science advances, “anomalies” (findings inconsistent with respect to established knowledge) should have begun a shift long ago in the paradigm of what he calls “normal science” [141]. Anomalies that challenge SMT are not new [28,99,142–145]. Puzzling findings at the organismal level predate the recent wave of hard-to-explain results produced by deep genome sequencing by more than half a century; they question the notion of cancer as a disease of the cell that is irreversibly transformed to a malignant phenotype by genetic alterations. On center stage is the ‘normalization’ of the cancerous cell accomplished by physical contact with a pertinent ‘embryonic field’ [146], which has prompted some researchers to view cancer as a problem of cellular differentiation [146–148].
“Normalization” is perhaps the most prosaic anomaly to the mutation paradigm. When Peter Nowell popularized clonal evolution of cancer cells [2], Beatrice Mintz’s group presented evidence that teratocarcinoma cells injected into mouse embryos (blastocysts) gave rise to cancer-free chimeric mice in which the mutated cancer cells were present in most organs. Such reversion of the malignant phenotype (i.e., normalization) within a proper tissue context has been reproduced in a variety of animal models [149]. Normalization is also observed during the ‘maturation’ of human tumors, whereby proliferating tumor cells sometimes develop into mature, non-proliferating cells, as most clearly seen in neuroblastoma, where cancer cells become ganglion cells [150,151]. In addition, some therapies, including target-selective inhibition of oncogenes aimed at killing cells, often trigger a wave of differentiation of the immature (mutated) cancer cells into specialized postmitotic cells, despite not being designed as differentiation therapy. This is most dramatically apparent in “differentiation syndrome”, the dreaded surge of differentiated neutrophils in the treatment of acute myeloid leukemia [152].
Thus, a latent developmental potential of cancer cells is occasionally realized, allowing the cells to (non-genetically) enter a mature state, provided a biological context that opens access to trajectories of physiological development. The old notion of cancer as a differentiation arrest is consistent with the potential for normalization of the malignant phenotype when the appropriate signals relieve the arrest [27,153–157]. Conversely, stressors, notably cytotoxic treatment itself, often cause ‘de-differentiation’ in the cells that survive treatment. As noted in point 10 of Table 1, cell stress can, independent of mutations, induce a stem-like state that confers xenobiotic resistance, reorganize the tumor microenvironment (TME), and promote a (pathological) healing by exploiting developmental fields, all in the absence of Darwinian selection of genetic mutants [158]. The logical inverse of Mintz’s experiments is the work that produced the teratocarcinoma cells used in those very experiments; these cells were generated by implanting normal embryos under the testicular capsule [159]. Later, it was observed that grafting normal (presumably mutation-free) embryonic stem cells into the mouse can produce teratocarcinoma [138].
These historical examples show that both differentiation (or ‘normalization’ if it happens in cancer cells) and de-differentiation (which creates the cancerous stem-like phenotype) are biological processes intimately linked to the existence of cancer. They represent drastic phenotype conversions that lack the characteristic of selective clonal expansion of mutated cells as the agent of pathological change. Instead, phenotypic transitions are controlled by a complex tissue context, which implies the involvement of an array of often poorly understood, coordinated signals.
The cell phenotype as collective actions of genes
A new malignant cell phenotype in tumor progression is still often explained by a genetic mutation that follows the conventional ‘one gene—one trait’ scheme. But, as widely noted, there is no simple correspondence (i.e., no 1:1 mapping) between genotype and phenotype [160]; genes directly code for proteins, not phenotypes, and they do not act alone. Gene loci interact with one another, via regulatory proteins that they encode, to collectively produce a cell phenotype [161,162]. The fundamental principle behind the fact that cells within an isogenic (clonal) cell population can have distinct phenotypes, as manifested in the existence of the cell types of the metazoan body, is that cells differ not in their genomes but in their genome-wide configuration of the activities of all gene loci. Such gene activity configurations produce the distinct gene expression patterns or transcriptomes that we can measure and interpret as the molecular basis of cell phenotypes.
The multiplicity of cell phenotypes given one same genome obviously defies the tacit assumption of a bijective mapping between genotype and phenotype. Such a relationship is required for Neo-Darwinian logic to work: natural selection enriches a population for a given genotype via a corresponding phenotype to which a fixed ‘fitness’ value is assigned that determines the selection advantage [55,112]. The phenotype diversification that produces the adult cell types from an unchanging genome follows the rules of development that are imposed by the regulatory interactions between the gene loci (and by interactions between the various cells in the tissue). The interactions between the genes are hard-wired in the genomic sequence and collectively form a complex network, the gene regulatory network (GRN), that orchestrates the activities of genes to produce those gene expression patterns that underlie the biologically meaningful phenotypes (Fig 1). Such a “genetic network” was first proposed by Jacob and Monod in the 1960s after they discovered that genes regulate other genes [163]. The central idea, as can be formulated mathematically, is that in coordinating the activities of genes, the GRN tends to interlock them in distinct, self-stabilizing gene expression configurations, called attractor states (valleys in Fig 1B). A key postulate, formulated in 1969 by Stuart Kauffman, is that attractors represent the gene expression patterns that correspond to cell types (or functional cell states) [162,164–166]. Being attractor states, such gene expression patterns are robust to perturbations; they re-establish spontaneously after disturbances, and thus can be inherited across cell generations. The existence of multiple attractor states is a feature of a particular class of complex dynamical systems to which GRNs belong [165].

Fig 1. The paradigm of cancer as a disease of gene regulation.
(A) The current paradigm of genetic mutation as the source of phenotypic change that drives tumorigenesis. (B) The new paradigm that explains cell phenotype changes by a concerted change of gene expression: the concept of the gene regulatory network (GRN) with gene-gene interaction mediated by transcription factors (TFs) or regulatory RNAs generalized a “wiring diagram”. It governs the dynamics of gene expression profiles that intuitively (Waddington, left) or with mathematical theory (right) can be represented as a landscape. In the landscape, the attractor states of the GRN, which define stable cell phenotypes, are the valleys. The theory predicts many more attractor states than can be occupied by cells in physiological states (cell types, in gray regions). The unused attractors (red) encode the gene expression profile thought to map into those of cancer cells.
Gene regulatory interactions of a given genome are implemented by transcription factors (TFs), and also by regulatory RNAs and chromosomal configurations. TF proteins and regulatory RNAs bind their targets in a sequence-specific manner. Thus, their regulatory action is highly gene locus-specific and is written in the genome; they collectively define the “wiring diagram” of the GRN (i.e., which gene regulates which genes, and how and under what conditions). The GRN was wired by evolution, e.g., via tuning of TF DNA binding motif sequences (trans-regulation) and their cognate target elements (cis-regulation). This resulted in binding specificities between regulators; such gene expression across all loci was forced into a set of genome-wide patterns, allowing the network to produce attractors (stable patterns) that encode meaningful cell phenotypes. The locus-specific TFs and regulatory RNAs are supported by the non-locus-specific apparatus of epigenetic modification (covalent marks on DNA and histone proteins) that sharpen signals and modulate temporal patterns [167]. Thus, the GRN is a distributed information processing system of mutually regulating genes that form a web of feedback loops—contrary to early views of genetic networks as hierarchical cascades of causation that rigidly link genotype to phenotype [168].
The GRN is not a clockwork. Stochasticity in cell-type diversification had already been proposed by Kupiec in 1983 [161]. Thermal fluctuations of molecule concentrations (due to the small number of molecules in cells) add a layer of non-determinism to the rules of the GRN, allowing attractor states to be represented as probabilities of being occupied [169–171]. This coexistence of deterministic logic and its probabilistic realization permits the definition of a potential-like quantity, which is aptly represented by Waddington’s epigenetic landscape, in which valleys correspond to attractor states and whose depth represent the relative stability of the cell phenotypes that they encode (Fig 2) [172,173]. (For simplicity, but sufficient for this discussion, we deal here only with “fixed-point attractors” and omit more complex dynamical structures, such as “limit cycles”, in which cells are trapped into a closed loop trajectory, such as the cell division cycle [174,175].)

Fig 2. Cancer attractors on the epigenetic landscape.
Mathematically, the epigenetic landscape can be extended to regions that represent gene activation configurations (in pink) that exist in theory but are never realized during development and in physiological tissues (green traces descending the hill in the gray regions). The unused regions (pink) contain unoccupied attractors—the cancer attractors. Under abnormal conditions cells can, upon massive perturbation of their gene expression profile, enter such cancer attractors that lack a path to the normal mature cell type attractors (bottom)—and thus are “maturation-arrested” and, if associated gene activation configurations are compatible with cell viability, can become cancerous. Mutations rewire the gene regulatory network (GRN), thus altering the dynamics, which can result in lowering of the barriers (hills) that prevent cells from leaving the physiological regions, facilitating the occupation of cancer attractors.
Now, here is a crucial corollary: on the epigenetic landscape of a given GRN, there exist, for mathematical and network-evolution reasons, many more attractor states than are occupied by cells in the healthy adult organism [176,177]. The phenotype of these unused attractors has been proposed to represent malignant cells [89,178]; in this view, cancer is a possibility immanent to metazoans, and hence is less suited to be seen as “being caused by something”, but rather is primarily the “unleashing of something latent”. Carcinogenesis would then be the accidental entry into these unused latently present attractors, which thus have been referred to as “cancer attractors” [178]. And these must be avoided by normal cells in development and under physiological conditions (Fig 2). This is the reason why the old adage of the molecular biologist that development is “tightly regulated” is more profound than one may think.
Waddington’s epigenetic landscape and cancer attractors
In the modern interpretation of the epigenetic landscape, cancer is thus a built-in but normally not realized feature, as epitomized by the unused attractors. Since, in physiological conditions, these attractors are unoccupied, they are not exposed as phenotypes and thus, they are ’not seen’ by natural selection; most of these states would establish gene expression patterns incompatible with life in the modern metazoan tissue. Those few unused attractors that can sustain viable cells in the tissue are not fine-tuned by selection. When occupied by cells that thus, by definition, become neoplastic, cancer attractors would command phenotypes with primordial behaviors not shaped by metazoan evolution to support cell societies that maintain tissue homeostasis [91,179]. In this perspective, cancer would represent atavism (“reversion to an ancestral form”): cancer cells would revive “selfish” protozoan functionalities, however imperfect [88,90,180,181]. More generally speaking, unused attractors represent cell “programs” of the phylogenetic past [182]. The accidental yet highly organized nature of vestigial phenotypes and their consistent reoccurrence is naturally explained by re-occupation of unused attractors that are an integral part of the same epigenetic landscape that is encoded by the genome and governs the unfolding of the normal extant phenotype. (The cancer atavism idea should, in its argumentation logic, not be conflated with the notion of proliferation and motility being the “default state” of cells (discussed in detail later) [91,179,183].)
Cancer cells are also stuck in an ontogenetic past. Because unused attractors do not partake in development of later evolved mature tissues, they are more likely to produce developmentally immature, dysfunctional traits that lack fine-tuned efficient homeostasis (mathematically represented by deep, smooth basins of attraction). For instance, being ‘unevolved’, they are likely to lack the sophisticated metazoan capacities to contribute to tissue morphogenesis or to replicate genomes with absolute fidelity. Most importantly, they are not connected by developmental paths to the mature cells’ attractor states (Fig 2, right). Such paths were carved by evolution (via GRN wiring) into the epigenetic landscape to ensure that normal immature cells respond to appropriate tissue signals during development and efficiently differentiate, while being shielded from undue entry into the unevolved cancer attractors (green curved arrows in Fig 2). Once trapped in such attractors of imperfect homeostasis, cells are easily perturbed by hypoxic, inflamed and or otherwise stressed stroma [184], all of which are known to cause genome and epigenomic instability, resulting in genetic alterations, further promoted by the absence of optimized DNA repair systems [185,186]. The immaturity of these cells places them closer to early developmental stages, in line with their expression of embryonic cell traits, such as accelerated cell division, multi-lineage potency, tolerance to genomic instability and xenobiotics—all features shared by cancer cells.
Having scant access to the physiological attractors of differentiated cell state [89] (Fig 2), cells in cancer attractors are also incapable of differentiation, hence the pathologists’ notion of maturation arrest [148]. The ‘accidental’ entry into cancer attractors does not depend on a specific mutation; instead, all kinds on non-genetic stressors that produce particular non-physiological, unstable gene expression patterns, amplified by stochastic fluctuations of transcriptomes, can trigger exit from normal cell type attractors and entry into a nearby cancer attractor (see point 10, Table 1).
Rethinking the concept of genetic causality for cancer
The concept of accidental entry of cells into a latently existing but undesired attractor and then being trapped in it, supports the notion of cancer as a process of unspecific “unleashing” (of a potential) rather than of specific “de novo causation”. Cancer attractors explain the unfathomable diversity of molecular disruptions and perturbations that all can accidentally trigger the entry into them and produce, as if orchestrated by an invisible hand, a specific gene activation configuration that governs the cancerous phenotype. These disruptions can result from an immense diversity of random genetic alterations that affect proteins of a variety of functional classes [21] or from a vast array of defects in regulatory interactions, from the rewiring of gene–gene interactions by genomic translocations (including creating novel fusion proteins or swapping promoters), the dysregulation of non-coding RNAs and RNA splicing, to altered function of broadly acting epigenetic and chromatin-modifying regulators. The immense diversity of mechanisms, all accidental, that can converge to producing the cancerous phenotype with minimal help by Darwinian selection, is perhaps the most prosaic yet profound manifestation of cancer attractors as latent structures in dark regions of the vast state space of gene activation patterns.
So, if genetic alterations are involved, what is our criticism of the claim of a genetic cause of cancer in the current paradigm? Cancer cannot be explained by explicit genetic causation in the traditional sense of “a gene X for that phenotype Y” being defective, as is readily achieved for mendelian diseases. Instead, we need to consider the collective action of genes, which we now can formalize with the epigenetic landscape, as well as cell population dynamics and tissue context. The landscape provides a formalism that places the role of a genetic anomaly for tumorigenesis in a new light: genetic mutations essentially alter the GRN wiring. In mathematical models that map the wiring diagram to the landscape topography, a localized GRN change (a point mutation or a larger scale rearrangement) is a localized rewiring and will most often just gently distort the topography of the landscape. There are only so many ways a landscape can gently change its shape (while still ensuring viability)—and one typical way is by altering the barrier height that separate the attractors. Such a distortion in turn affects the cell trajectories. For instance, it could facilitate the accidental entry into a cancer attractor from a given place in the normal trajectory, driven by molecular noise or non-mutagenic environmental stress (details in [55,158]). It is in this convoluted sense that one can admit that genetic mutations can “cause” cancer.
Despite the absence of developmental paths to normal mature states, the epigenetic landscape still grants cells in cancer attractors an intrinsic potential for normalization, as manifested in the rare reversion of the cancerous phenotype under particular experimental conditions [153,155,156,187,188]. The reversion potential is rarely realized because very distinct tissue constellations are required to overcome the energy barrier to exit the cancer attractor, as seen in the particular instance of neuroblastoma that can spontaneously normalize. The difficulty of normalization is consistent with the limited success of differentiation therapy [189,190] and with the exceeding rarity of “spontaneous remission” of cancer despite the theoretical reversibility of cell states [191].
We can now more precisely articulate an indirect, convoluted role of genetic mutations in cancer to reconcile with the SMT view. Both the genetic paradigm of cancer and Neo-Darwinism rest on the tacit assumption of a 1:1 mapping between genotype and phenotype [192]. In this view, one must, by logical necessity, explain any phenotypic change by a genetic mutation. But, as computer simulations have suggested, most mutations in the core GRN (which in principle alter the wiring of the GRN) do not qualitatively affect the topography of the epigenetic landscape (e.g., they typically do not destroy or create attractor states), but as said above, cause a gentle distortion. Thus, the majority of mutations, even those that alter gene activity, are readily buffered away. However, mild distortions due to GRN rewiring can alter the relative stabilities of attractors, and thus may affect transition rates between attractors (e.g., stem cell and differentiated state and senescent states), including rates for epithelial-mesenchymal transitions (EMT)[193,194] and entry to the unused attractors [195,196]. Altered transition rates in turn lead to imbalances in tissue homeostasis. One manifestation is the widely observed trans-differentiation event (lineage switching, “lineage infidelity”) seen in malignancy.
As to the much-needed higher-level organismal biology view of cancer as a complex disease, a first step towards organicism is in the interpretation of large-scale genomic alterations as the source of cell physiological and not genetic informational departure from the norm. Aneuploidy and polyploidy in cancer cells [197] often result in the formation of polyploid giant cancer cells that are found in nearly half of all high-grade cancers [198]. Polyploidy and associated expression of meiotic genes in malignant cells offer a link between cancer biology and organismal biology at a larger timescale and could represent a rather generic alternative attractor that is often accessed by tumorigenic dysregulation of the GRN. In fact, since tumors often reactivate diapause and gametogenesis programs that mark the entry into a new sexual reproduction cycle and epitomize the immortality of the germline, it has been hypothesized that cancer may recapitulate, albeit in an aberrant and abortive manner, the life cycle of organisms [180,199,200].
Finally, beyond its putative role in generating mutations (and neoantigens) [201], there are also organismal consequences of genomic instability (the “mutator phenotype”), which itself is more likely a consequence of the abnormal phenotype produced by unevolved attractors than the needed source of cancer-causing mutations. Indeed, the idea of what actually “came first” was articulated by Richmond Prehn: “…it may be more correct to say that cancers beget mutations than it is to say that mutations beget cancers” [202]. Then, we shall also remember the physical, not informational consequences of genome instability: the DNA repair response triggered by genomic instabilities produces unphysiological DNA configurations (breakpoints, stalled replication forks, RNA–DNA hybrids, double- and single-stranded DNA, etc.) that leak into the cytoplasm, which in turn, mistaken for viral invasion, activates stress and inflammatory responses via the NFkB and STING/interferon pathways, thus affecting the cell’s microenvironment [203,204]. This brings us to the tissue level processes.
Cancer as a tissue-based disease: The tissue organization field theory of carcinogenesis
The interpretation of genome sequence-based lineage reconstruction (Table 1, point 6) defaults to the view that the tumor originates from a single cell, the “initiating” or “founder”, cancer cell. This is correct in a fundamental, almost trivial way because a group of cells always has a common ancestor—it just depends on how far back one traces the pedigree (up to the zygote). The idea of one initiating cell in which the truncal (“tumor initiating”) mutation in the clonal tree happened, making it a founder cell [36], ignores that its outgrowth has to depend on its neighboring cells (normal or transformed) that provide the critical tissue context for it to survive and also ignores the multiplicity of distinct cell phenotypes within an isogenic clone. Conversely, not all cells that contain that same early mutation (that would allow for later oncogenic, otherwise lethal mutations to accumulate) will become cancerous—see Table 1, points 6 and 7 [108].
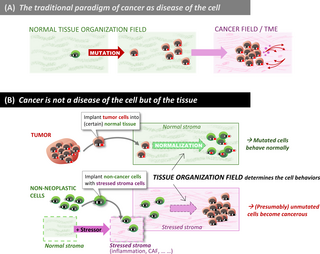
Empirical evidence contradicting the view of “cancer as a disease of the cell” was already available over 60 years ago, when David W. Smithers published an essay entitled “An Attack to Cytologism” [205]. Smithers refuted the idea that cancer originates from an abnormal cell. His and others’ observations suggested that cancer is not a disease that starts from a single tumor-initiating cell, but rather an anomaly of tissue organization (Fig 3). (This argument only proposes that it is not as simple as postulating “a” cancer cell but does not deny the existence of individual cells with enhanced potential to drive tumor growth).

Fig 3. Cancer as a disease of the tissue, not the cell.
(A) The paradigm of abnormal cells as the source of tumors. (B) The new paradigm of cancer as a disease of the tissue, explained by schematics of seminal experiments that establish the phenomenon of normalization of cancerous cells by the stroma, as well as the converse, i.e., the initiation of tumor from “healthy cells” due to carcinogenic stressors acting on the stroma. Note that this second paradigm shift implicitly requires non-genetic plasticity of cancer cells, as proposed in the first paradigm shift (Fig 1). In the traditional paradigm, plasticity is assumed only for the (non-transformed) stromal cells.
A compelling Illustration of the need for a tissue-based rather than cell-centric view is the finding that while a bulk injection of hepatocarcinoma cells into the liver generates a liver tumor, injection of the same amounts of the same cell type into the spleen, which distributes transplanted cells individually throughout the liver, fails to generate a tumor [153]. Importantly, the cells injected into the spleen became normalized in the liver by direct contact with their neighboring normal hepatic cells and were incorporated into the liver’s normal structure, underscoring the role of non-genetic, environment-regulated cell phenotype plasticity at the demarcation between normalcy and neoplasia [154,206]. These observations led to the concept that there is no “cancer cell” [101] because cells declared as such (e.g., because of canonical oncogenic mutations) behave as normal cells (Table 1, Point 2).
Instead, cancer can be broadly understood as “development gone awry”. Within this perspective, the tissue organization field theory is based on two principles that unite phylogenesis and ontogenesis. Firstly, the default state of all cells is constitutive proliferation with variation and motility. Therefore, proliferation and motility do not require an explanation; instead, what ought to be explained is why cells do not proliferate and move. Secondly, cancer is a tissue-based disease, whereby the tissue organization constraints to the default state of its cells are weakened. Consequently, cells become freed to express their default state, thus proliferating, generating variation and moving. This explains tumor growth by accrual of new cells, as well as invasion and metastasis [91,102].
The other major facet of carcinogenesis as a tissue-level phenomenon pertains to the role of ‘non-cancerous’ cells in the TME or the stroma. The idea that the TME, consisting of extracellular matrix (ECM), mesenchymal cells, and resident and invading immune cells, plays a role in carcinogenesis was accepted by the cancer community only in the 1990s, despite pointed reference by the pathologist Rudolph Virchow more than 150 years ago [207]. The all too obvious role of the TME in carcinogenesis, which involves angiogenesis, inflammation, immune suppression and cancer-associated fibroblasts (CAFs) [208], soon led to exploding research funding in this area. However, the stromal component has mostly been seen as a necessary enabler and rarely as a driver of carcinogenesis. It provided the biological embodiment of the convenient notion of context that is often invoked to mitigate anomalies plaguing the SMT. Throughout the 20th century, carcinogenesis was considered a genetic disease of the cancer cell despite accumulating experimental evidence pointing to a causative role of the stroma [209]. For example, the fact that injection of a chemical carcinogen administered intraperitoneally generates mammary cancers in susceptible rat strains has been attributed to mutations caused by the carcinogen in the glandular epithelial cells, such as Ha-ras-1 (details below) [210]. However, all cells of the organism are exposed to the carcinogen. From a developmental biology perspective, the question is, which is the target of the carcinogen: the epithelium, the stroma or both?
Lessons from organogenesis
To understand the relationship between the stroma (the “support” scaffold of an organ) and the parenchyma (the “functional” part of the organ) in cancer, we shall review the process of organogenesis [211,212]. The genesis of tissues and organs entails complex reciprocal interactions between the components of the morphogenetic field defined by a distinct 3D array of cells that gives rise to an autonomous tissue structure or organ [213]. Numerous epithelium/mesenchyme recombination experiments have shown that it is the mesenchyme that determines the type of skin adnexa; for example, wing mesenchyme dictates the presence of chicken feathers regardless of whether it is recombined with epidermis from foot or wing, and scales appear regardless of the origin of the epidermis if recombined with mesenchyme of the foot [214]. Similarly, the branching pattern of the epithelium of salivary and mammary glands is determined by the stroma of the organ [215].
As in organogenesis, the stroma is equally as important in tumorigenesis. When mammary epithelial cells were exposed in vitro either to the mutagenic carcinogen N-nitrosomethylurea (NMU), or to vehicle before being transplanted into the mammary stroma of rats surgically cleared of epithelium [145,183], carcinogenesis only occurred when the stroma was exposed in vivo to NMU, regardless of whether or not the epithelial cells were exposed to the carcinogen (Fig 3). Mammary epithelial cells exposed in vitro to the carcinogen formed phenotypically normal ducts when injected into unexposed stroma. Mutations in the Ha-ras-1 gene, which was assumed to be the cause of cancerization [210], did not correlate with initiation of neoplasia: not only was it often found in both cleared mammary fat pads of vehicle-treated animals and intact mammary glands of untreated animals, but it was also absent in various carcinomas. Such tissue recombination experiments show that the stroma is a crucial target of the carcinogen, while the epithelium is not, and that in this case, mutation in the Ha-ras-1 gene is neither necessary nor sufficient for neoplastic development [216]. A comparable experiment showed that radiation-induced changes in mammary gland stroma contributed to the neoplastic progression of non-irradiated, quasi-normal (TP53-mutated but non-tumorigenic) mammary epithelial cells [217]. Finally, nontumorigenic human prostatic epithelial cells from benign prostate hyperplasia develops into tumors when recombined with prostate cancer stroma and not with normal stroma [218,219]. Recent stromal-epithelial tissue recombinant culture systems and single-cell transcriptomics have begun to unravel the molecular signals that mediate such stromal tumorigenic effects [220].
Not only can carcinogenesis be initiated by injuring the stroma, but a normal stroma can also act as a gate-keeper of neoplastic development by constraining cells belonging to a neoplasm to behave as the normal cells of the organ that form normal tissue structures [221]. The experimental normalization of mutated cells that were once part of a tumor has been reported for a variety of cancer types, among them melanoma (by the neural crest [222]), hepatocarcinoma (by normal liver [223]), or mammary cancer (by normal mammary gland [27,221,224–226]). A compelling demonstration of a tumor-restraining role of stroma is the case of localized pancreatic cancer, where targeting of (originally considered tumor-promoting) CAFs resulted in tumor progression and invasion [227–229].
Intriguingly, the same chemically inert material can cause tumors in animals when implanted as rods but not when injected as powder of the same material and same mass [230]. This suggests that chronic wounding and/or mechanical stress of the tissue causes carcinogenesis through breakdown of physical tissue architecture in the absence of genotoxicity, something that has long been observed in multiple cancer models, underscoring the importance of tissue fields [231–234]. In the light of the central role of the mesenchyme on the determination of the parenchyma phenotype, it is unsurprising to see the surge of data showing a tumor-promoting effect of altered stroma, most notably, ECM composition, fibroblast phenotype (in the form of CAFs) and tumor-promoting chronic inflammation [209]. While these findings have triggered a massive quest for underlying chemical mediators, one should not forget that tissue organization plays not just a supporting, but a determinant role.
The organicist perspective [138] is based on the interdependency of the organism and its organs. It recognizes a circular causal regimen by closure of constraints that makes parts interdependent [235], wherein these constraints are not only molecules, but also biophysical force [236]. Since such a view encompasses tissues not simply as collectives of cells but as emergent anatomical and functional organized entities, it affords little opportunity for discrete molecular targeting, limiting its appeal to reductionist research programs in search of a molecular ‘silver bullet’.
Explaining anomalies in cancer biology under the SMT paradigm
A common way of explaining the lack of fit between the SMT and data has been to invoke involvement of biological processes not obviously related to oncogenic mutations, such as cellular metabolism, stromal alterations, bacterial and viral carcinogenesis, immune surveillance, etc. that we have characterized as “compromises”. In such hybridization of explanations, old ideas are kept intact, such as the claim that cancer is a disease of the cell and that a normal cell is rendered cancerous by genetic alterations. Justifications often include epimutations [237] due to altered epigenetic marks (epigenetic reprogramming) or references to the old dualism between initiator (mutagenic) and promoter (non-mutagenic) [238].
The concept of dividing the cause of cancer into the initiator, which is the mutagenic event, and the promoter, a chronic (chemical) stress by typically non-genotoxic agents, has been proposed based on the classical experiments of skin carcinogenesis, first performed a century ago. Mice subjected to chemicals known to be mutagenic will develop skin cancer only after ensuing repeated treatment with a non-genotoxic irritant, such as croton oil or the phorbol ester 12-O-tetra-decanoylphorbol-13-acetate (TPA) [192]. Recent genomic sequence analyses reveal that the initial carcinogen indeed caused genetic alterations, which remain silent until application of TPA at a later time point. After TPA treatment, development of papillomas and carcinomas was observed [239]. The elegance of the concept in its modern reincarnation is that the promoter is now understood to not only exert its effect on the mutated cells via non-genetic phenotype switching, but also on the TME via tumorigenic inflammation. The initiator-promoter model is often used to explain the well-known fact that a large fraction of listed carcinogens is non-mutagenic; they may apparently cause cancer by triggering outgrowth of occult mutated cancerous cells. However, this picture is still too simplistic, exemplifying the aforementioned hybridization of old ideas with new findings. At its core, it still maintains a primary causative role of genetic mutations in a single cancer initiating cell, followed by a sequence of causal steps. The qualitative dichotomy between a mutagenic initiator that creates ’cancer cells’ and the non-genetic, tissue-perturbing promoter that expands them may not be as clear-cut. Indeed, the reverse experiment (first treatment with the promoter followed by the initiator) equally produces tumors [240]. This result refutes the classical model that requires that the mutagenic (alleged) initiator must act first. Instead, the reverse experiment suggests merely a synergism between initiator and promoter, albeit a complex one that must involve tissue memory of non-mutagenic perturbations. The promoter/initiator framework by itself is an interesting model, but further investigation should be conducted through the lens of non-genetic dynamics, tissue organization (including the TME) and organismal biology.
Another aspect that has been invoked to alleviate the discrepancies between genome sequencing results and the SMT is epigenetic reprogramming [241]. A solid, formal explanation for enduring changes in the landscape of cell phenotype would have to consider the dynamics of GRNs because epigenetic marks themselves are not explanatory; they are reversible, and the epigenetic modifiers that place these marks are not locus-specific and must be guided by the GRN to coordinate gene loci activities to produce coherent expression patterns that can be locked-in (attractors) [162,167]. Moreover, the attractor stability of a cell phenotype can be disrupted by drastic changes in its microenvironment that affect a large number of regulatory genes to overcome attractor dynamics. This property is evident in the recombination studies with fetal mesenchyme, underscoring the strong leverage that tissue level interactions and tissue architecture possess in perturbing the GRN at multiple points to trigger attractor destabilization and transitions [231,242,243].
Interestingly, the hybridization of concepts to accommodate anomalies is also present in the extended list of the ‘Hallmarks of cancer’. Indeed, hallmarks only serve as attachment points for tenuous molecular causes, dictated by Dennett’s “greedy” reductionism, namely the recourse by scientists to “underestimate the complexities, trying to skip whole layers or levels of theory in their rush to fasten everything securely and neatly to the foundation” [206]. This attitude became evident in the latest installment of the “Hallmarks of cancer” listing [111] that, instead of biological thinking, uses the existence of mutated genetic pathways to motivate new hallmarks that by necessity are explained by them. Notwithstanding, the two new hallmarks, “Unlocking phenotypic plasticity” and “Non-mutational epigenetic programming”, are non-genetic, with ironic redundancy between them [111].
Conclusions and outlook
Dealing with a complex world, philosophers of science talk about auxiliary hypotheses, which are readily erected using known facts. These abound because complex systems contain a web of causal interactions, as epitomized by the living organism. They offer an uncountable set of auxiliary explanations that can rescue any existing paradigm and not only renders the falsification of a hypothesis meaningless [244,245] but also its defense by excuse arguments. Put differently, the genetic paradigm of cancer is a vague theory and, as the physicist Richard Feynman stated, vague theories cannot be disproven [246], which is even more true for as complex a processes as carcinogenesis. Therefore, interpretation of empirical results guided by formal theory based on some set of first principles, and undaunted by dogmas, is ever more important in an era of easy data acquisition.
Over the years, many proposals outside the paradigm of cancer as a genetic disease have been offered in view of the accumulating findings pointing to shortcomings of the SMT. Some scientists in the SMT camp—still a minority—now begin to embrace a pluralistic view. For instance, we must recall Mike Stratton’s quote in the opening section of this Essay. After his team failed to find mutational signatures specific to endemic squamous cell carcinoma of the esophagus, he concluded:
“We have found evidence that chemicals might be able to work in different ways other than directly causing mutations to increase a person’s chances of developing cancer. We will have to rethink our ideas about the way in which some cancers develop. It is a crucial lesson” [205].
We hope that such “crucial lessons” will not be ignored. Pursuing a rigorous explanation of carcinogenesis entails identification of fundamental biological principles placed within a consistent formal theoretical framework, instead of more terminological acrobatics centered around observed altered molecular pathways. As Darwin pointed out to a friend in 1821,
if one were “only to observe, not theorise… [one] might as well go into a gravel-pit and count the pebbles and describe their colours. How odd it is that everyone should not see that all observation must be for or against some view, if it is to be of any service” [247].
Theories do not need to be right; they are a practical tool to conduct research. If Darwin’s pebbles are today’s molecular alterations in tumors, then we must not simply be satisfied with their categorization in a system of ingredients that cause tumors, as illustrated by the proposed hallmarks. We must rather regard the hallmarks merely as manifestations of fundamental principles of living organisms. An epistemic shift towards a biological theory of cancer may still be an uphill battle in the current climate of thought created by the ease of data collection and a culture of research that discourages ’disruptive science’ [248]. Here, we have made an argument for dropping the SMT and its epicycles. We presented new and old but sidelined theoretical alternatives to the SMT that embrace theory and organismal biology and can guide experiments and data interpretation. We expect that the diminishing returns from the ceaselessly growing databases of somatic mutations, the equivalent to Darwin’s gravel pit, may soon reach a pivot point. In addition, we hope that then the adherents of the genetic paradigm of cancer will begin to expand their vista on cancer in a profound way.
Acknowledgments
The authors would like to thank our long-time colleagues for countless inspiring discussions, including Michel Aguet, Donald Ingber, Stuart Kauffman, Giuseppe Longo, Maël Montévil, Matteo Mossio, Ilya Shmulevich, Thea Tlsty and Yojiro Yamanaka. We also would like to thank Cheryl Schaeberle and Victoria Bouffard for their critical reading of the manuscript. The landscape topography of Fig 2 was generated with help of the artificial intelligence tool Microsoft Copilot Designer (DALLE-3) using a surface wave texture obtained from freepik.com
References
- 1.
Cairns J. Mutation selection and the natural history of cancer. Nature. 1975;255(5505):197–200. pmid:1143315 - 2.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8. pmid:959840 - 3.
Weinberg RA. Leaving home early: reexamination of the canonical models of tumor progression. Cancer Cell. 2008;14(4):283–4. pmid:18835030 - 4.
Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9(4):138–41. pmid:8516849 - 5.
Weinberg RA. Coming full circle-from endless complexity to simplicity and back again. Cell. 2014;157(1):267–71. pmid:24679541 - 6.
Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24. pmid:19360079 - 7.
West HJ. No solid evidence, only hollow argument for universal tumor sequencing: show me the data. JAMA Oncol. 2016;2(6):717–8. pmid:27078630 - 8.
Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev. 2014;24(100):52–60. pmid:24657537 - 9.
Prasad V, Fojo T, Brada M. Precision oncology: origins, optimism, and potential. Lancet Oncol. 2016;17(2):e81–6. pmid:26868357 - 10.
Rosenzweig SA. Acquired resistance to drugs targeting receptor tyrosine kinases. Biochem Pharmacol. 2012;83(8):1041–8. pmid:22227013 - 11.
Sanchez-Laorden B, Viros A, Girotti MR, Pedersen M, Saturno G, Zambon A, et al. BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal. 2014;7(318):ra30. pmid:24667377 - 12.
Brock A, Huang S. Precision oncology: between vaguely right and precisely wrong. Cancer Res. 2017;77(23):6473–9. pmid:29162615 - 13.
Michaeli DT, Michaeli T. Overall survival, progression-free survival, and tumor response benefit supporting initial us food and drug administration approval and indication extension of new cancer drugs, 2003-2021. J Clin Oncol. 2022;40(35):4095–106. pmid:35921606 - 14.
Marquart J, Chen EY, Prasad V. Estimation of the Percentage of US patients with cancer who benefit from genome-driven oncology. JAMA Oncol. 2018;4(8):1093–8. pmid:29710180 - 15.
Tannock IF, Hickman JA. Limits to personalized cancer medicine. N Engl J Med. 2016;375(13):1289–94. pmid:27682039 - 16.
Versteeg R. Cancer: tumours outside the mutation box. Nature. 2014;506(7489):438–9. pmid:24553138 - 17.
Moore R. Niels Bohr: the man and the scientist: Hodder & Stoughton; 1967. - 18.
Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stütz AM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–50. pmid:24553142 - 19.
Ohnishi K, Semi K, Yamamoto T, Shimizu M, Tanaka A, Mitsunaga K, et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156(4):663–77. pmid:24529372 - 20.
Moody S, Senkin S, Islam SMA, Wang J, Nasrollahzadeh D, Cortez Cardoso Penha R, et al. Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat Genet. 2021;53(11):1553–63. pmid:34663923 - 21.
de Magalhães JP. Every gene can (and possibly will) be associated with cancer. Trends Genet. 2022;38(3):216–7. pmid:34756472 - 22.
Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880–6. pmid:25999502 - 23.
Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. pmid:18167340 - 24.
Kato S, Lippman SM, Flaherty KT, Kurzrock R. The conundrum of genetic “Drivers” in benign conditions. J Natl Cancer Inst. 2016;108(8):djw036. pmid:27059373 - 25.
Williams N, Lee J, Mitchell E, Moore L, Baxter EJ, Hewinson J, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature. 2022;602(7895):162–8. pmid:35058638 - 26.
Li R, Du Y, Chen Z, Xu D, Lin T, Jin S, et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science. 2020;370(6512):82–9. pmid:33004515 - 27.
Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? Bioessays. 2006;28(5):515–24. pmid:16615084 - 28.
Baker SG, Kramer BS. Paradoxes in carcinogenesis: new opportunities for research directions. BMC Cancer. 2007;7:151. pmid:17683619 - 29.
Ling G, Persson A, Berne B, Uhlén M, Lundeberg J, Ponten F. Persistent p53 mutations in single cells from normal human skin. Am J Pathol. 2001;159(4):1247–53. pmid:11583952 - 30.
Weaver JMJ, Ross-Innes CS, Shannon N, Lynch AG, Forshew T, Barbera M, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet. 2014;46(8):837–43. pmid:24952744 - 31.
Kim SY, Jung S-H, Kim MS, Baek I-P, Lee SH, Kim T-M, et al. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6(10):7597–607. pmid:25831047 - 32.
Hafner C, López-Knowles E, Luis NM, Toll A, Baselga E, Fernández-Casado A, et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc Natl Acad Sci U S A. 2007;104(33):13450–4. pmid:17673550 - 33.
Tschandl P, Berghoff AS, Preusser M, Burgstaller-Muehlbacher S, Pehamberger H, Okamoto I, et al. NRAS and BRAF mutations in melanoma-associated nevi and uninvolved nevi. PLoS One. 2013;8(7):e69639. pmid:23861977 - 34.
Baverstock K, Karotki AV. Towards a unifying theory of late stochastic effects of ionizing radiation. Mutat Res. 2011;718(1–2):1–9. pmid:21078408 - 35.
Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–10. pmid:22237025 - 36.
Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nat Genet. 2015;47(3):209–16. pmid:25665006 - 37.
Ling S, Hu Z, Yang Z, Yang F, Li Y, Lin P, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc Natl Acad Sci U S A. 2015;112(47):E6496-505. pmid:26561581 - 38.
Robertson-Tessi M, Anderson ARA. Big Bang and context-driven collapse. Nat Genet. 2015;47(3):196–7. pmid:25711865 - 39.
Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nat Genet. 2016;48(3):238–44. pmid:26780609 - 40.
Bakhoum SF, Landau DA. Cancer evolution: no room for negative selection. Cell. 2017;171(5):987–9. pmid:29149612 - 41.
Yang Y, Li S, Wang Y, Zhao Y, Li Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct Target Ther. 2022;7(1):329. pmid:36115852 - 42.
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46(3):225–33. pmid:24487277 - 43.
Notta F, Mullighan CG, Wang JCY, Poeppl A, Doulatov S, Phillips LA, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469(7330):362–7. pmid:21248843 - 44.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–33. pmid:24522528 - 45.
Morrissy AS, Garzia L, Shih DJH, Zuyderduyn S, Huang X, Skowron P, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016;529(7586):351–7. pmid:26760213 - 46.
Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta Rev Cancer. 2017;1867(2):151–61. pmid:28110020 - 47.
Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805(1):105–17. pmid:19931353 - 48.
Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477–82. pmid:33116311 - 49.
Nam AS, Kim K-T, Chaligne R, Izzo F, Ang C, Taylor J, et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature. 2019;571(7765):355–60. pmid:31270458 - 50.
Velten L, Story BA, Hernández-Malmierca P, Raffel S, Leonce DR, Milbank J, et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat Commun. 2021;12(1):1366. pmid:33649320 - 51.
Lee SE, Kang SY, Yoo HY, Kim SJ, Kim WS, Ko YH. Clonal relationships in recurrent B-cell lymphomas. Oncotarget. 2016;7(11):12359–71. pmid:26848863 - 52.
Oren Y, Tsabar M, Cuoco MS, Amir-Zilberstein L, Cabanos HF, Hütter J-C, et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021;596(7873):576–82. pmid:34381210 - 53.
Williams MJ, Oliphant MUJ, Au V, Liu C, Baril C, O’Flanagan C, et al. Luminal breast epithelial cells of BRCA1 or BRCA2 mutation carriers and noncarriers harbor common breast cancer copy number alterations. Nat Genet. 2024;56(12):2753–62. pmid:39567747 - 54.
Lips EH, Kumar T, Megalios A, Visser LL, Sheinman M, Fortunato A, et al. Genomic analysis defines clonal relationships of ductal carcinoma in situ and recurrent invasive breast cancer. Nat Genet. 2022;54(6):850–60. pmid:35681052 - 55.
Huang S. Reconciling non-genetic plasticity with somatic evolution in cancer. Trends Cancer. 2021;7(4):309–22. pmid:33536158 - 56.
Gopalan V, Singh A, Rashidi Mehrabadi F, Wang L, Ruppin E, Arda HE, et al. A transcriptionally distinct subpopulation of healthy acinar cells exhibit features of pancreatic progenitors and PDAC. Cancer Res. 2021;81(15):3958–70. pmid:34049974 - 57.
Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. 2008;453(7194):544–7. pmid:18497826 - 58.
Gutierrez C, Al’Khafaji AM, Brenner E, Johnson KE, Gohil SH, Lin Z, et al. Multifunctional barcoding with ClonMapper enables high-resolution study of clonal dynamics during tumor evolution and treatment. Nat Cancer. 2021;2(7):758–72. pmid:34939038 - 59.
Takahashi N, Miura I, Saitoh K, Miura AB. Lineage involvement of stem cells bearing the Philadelphia chromosome in chronic myeloid leukemia in the chronic phase as shown by a combination of fluorescence-activated cell sorting and fluorescence in situ hybridization. Blood. 1998;92(12):4758–63. pmid:9845542 - 60.
Shlyakhtina Y, Bloechl B, Moran K, Maslakova A, Munro A, Carragher N, et al. Cytoplasmic lncRNAs nucleate signalling pathways to define metastable state dynamics and determine phenotypic output. 2024. - 61.
Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. 2004;305(5690):1622–5. pmid:15308767 - 62.
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108(19):7950–5. pmid:21498687 - 63.
Brock A, Chang H, Huang S. Non-genetic heterogeneity—a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. 2009;10(5):336–42. pmid:19337290 - 64.
Yano S, Nakataki E, Ohtsuka S, Inayama M, Tomimoto H, Edakuni N, et al. Retreatment of lung adenocarcinoma patients with gefitinib who had experienced favorable results from their initial treatment with this selective epidermal growth factor receptor inhibitor: a report of three cases. Oncol Res. 2005;15(2):107–11. pmid:16119008 - 65.
Pisco AO, Brock A, Zhou J, Moor A, Mojtahedi M, Jackson D, et al. Non-Darwinian dynamics in therapy-induced cancer drug resistance. Nat Commun. 2013;42467. pmid:24045430 - 66.
Ghisolfi L, Keates AC, Hu X, Lee D, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012;7(8):e43628. pmid:22928007 - 67.
Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J, et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci Transl Med. 2017;9(397):eaan0026. pmid:28679654 - 68.
Nör C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, et al. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. 2014;16(2):137–46. pmid:24709421 - 69.
Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546(7658):431–5. pmid:28607484 - 70.
Su Y, Wei W, Robert L, Xue M, Tsoi J, Garcia-Diaz A, et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A. 2017;114(52):13679–84. pmid:29229836 - 71.
Marin-Bejar O, Rogiers A, Dewaele M, Femel J, Karras P, Pozniak J, et al. Evolutionary predictability of genetic versus nongenetic resistance to anticancer drugs in melanoma. Cancer Cell. 2021;39(8):1135-1149.e8. pmid:34143978 - 72.
Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182(5):1232–1251.e22. pmid:32822576 - 73.
Hung T-H, Hsu S-C, Cheng C-Y, Choo K-B, Tseng C-P, Chen T-C, et al. Wnt5A regulates ABCB1 expression in multidrug-resistant cancer cells through activation of the non-canonical PKA/β-catenin pathway. Oncotarget. 2014;5(23):12273–90. pmid:25401518 - 74.
Sun Y, Campisi J, Higano C, Beer TM, Porter P, Coleman I, et al. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med. 2012;18(9):1359–68. pmid:22863786 - 75.
Lee E, Yang J, Ku M, Kim NH, Park Y, Park CB, et al. Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell Death Dis. 2015;6(7):e1805. pmid:26136078 - 76.
Zhang M, Zhang YY, Chen Y, Wang J, Wang Q, Lu H. TGF-β signaling and resistance to cancer therapy. Front Cell Dev Biol. 2021;9:786728. pmid:34917620 - 77.
Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: “What does not kill me strengthens me”. Br J Cancer. 2015;112(11):1725–32. pmid:25965164 - 78.
Bell CC, Gilan O. Principles and mechanisms of non-genetic resistance in cancer. Br J Cancer. 2020;122(4):465–72. pmid:31831859 - 79.
Marine J-C, Dawson S-J, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. 2020;20(12):743–56. pmid:33033407 - 80.
Salgia R, Kulkarni P. The genetic/non-genetic duality of drug “resistance” in cancer. Trends Cancer. 2018;4(2):110–8. pmid:29458961 - 81.
Smith LK, Sheppard KE, McArthur GA. Is resistance to targeted therapy in cancer inevitable?. Cancer Cell. 2021;39(8):1047–9. pmid:34375607 - 82.
National Cancer Institute. The Cancer Genome Atlas (TCGA). Available from: http://cancergenome.nih.gov/abouttcga/overview. - 83.
Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science. 2020;370(6512):75–82. pmid:33004514 - 84.
Rubin H. Cancer as a dynamic developmental disorder. Cancer Res. 1985;45(7):2935–42. pmid:3891078 - 85.
Xie K, Abbruzzese JL. Developmental biology informs cancer: the emerging role of the hedgehog signaling pathway in upper gastrointestinal cancers. Cancer Cell. 2003;4(4):245–7. pmid:14585350 - 86.
Lee K-H, Li M, Michalowski AM, Zhang X, Liao H, Chen L, et al. A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc Natl Acad Sci U S A. 2010;107(1):69–74. pmid:20018659 - 87.
Domazet-Loso T, Tautz D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010;8:66. pmid:20492640 - 88.
Davies PCW, Lineweaver CH. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys Biol. 2011;8(1):015001. pmid:21301065 - 89.
Huang S. On the intrinsic inevitability of cancer: from foetal to fatal attraction. Semin Cancer Biol. 2011;21(3):183–99. pmid:21640825 - 90.
Zhou JX, Cisneros L, Knijnenburg T, Trachana K, Davies P, Huang S. Phylostratigraphic analysis of tumor and developmental transcriptomes reveals relationship between oncogenesis, phylogenesis and ontogenesis. Converg Sci Phys Oncol. 2018;4(2):025002. - 91.
Sonnenschein C, Soto AM. The society of cells: cancer and control of cell proliferation: Garland Science; 1998. - 92.
Wagner GP, , Dighe A, Levchenko A. The coevolution of placentation and cancer. Annu Rev Anim Biosci. 2022;10:259–79. pmid:34780249 - 93.
Monti N, Verna R, Piombarolo A, Querqui A, Bizzarri M, Fedeli V. Paradoxical behavior of oncogenes undermines the somatic mutation theory. Biomolecules. 2022;12(5):662. pmid:35625590 - 94.
Frank SA, Yanai I. The origin of novel traits in cancer. Trends Cancer. 2024;10(10):880–92. pmid:39112299 - 95.
Sherr CJ. Cancer cell cycles. Science. 1996;274(5293):1672–7. pmid:8939849 - 96.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. pmid:22258609 - 97.
Vaux DL. In defense of the somatic mutation theory of cancer. Bioessays. 2011;33(5):341–3. pmid:21503936 - 98.
Nordling CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7(1):68–72. pmid:13051507 - 99.
Brücher BLDM, Jamall IS. Somatic mutation theory—why it’s wrong for most cancers. Cell Physiol Biochem. 2016;38(5):1663–80. pmid:27160408 - 100.
Nowak MA, Michor F, Iwasa Y. Genetic instability and clonal expansion. J Theor Biol. 2006;241(1):26–32. pmid:16405914 - 101.
Sonnenschein C, Soto AM. The death of the cancer cell. Cancer Res. 2011;71(13):4334–7. pmid:21507929 - 102.
Soto AM, Longo G, Montévil M, Sonnenschein C. The biological default state of cell proliferation with variation and motility, a fundamental principle for a theory of organisms. Prog Biophys Mol Biol. 2016;122(1):16–23. pmid:27381480 - 103.
Evans JA, McDonald SAC. The complex, clonal, and controversial nature of Barrett’s esophagus. Adv Exp Med Biol. 2016;908:27–40. pmid:27573766 - 104.
Kakiuchi N, Ogawa S. Clonal expansion in non-cancer tissues. Nat Rev Cancer. 2021;21(4):239–56. pmid:33627798 - 105.
Majeti R, Jamieson C, Pang WW, Jaiswal S, Leeper NJ, Wernig G, et al. Clonal expansion of stem/progenitor cells in cancer, fibrotic diseases, and atherosclerosis, and CD47 protection of pathogenic cells. Annu Rev Med. 2022;73:307–20. pmid:35084991 - 106.
Kang L, He G, Sharp AK, Wang X, Brown AM, Michalak P, et al. A selective sweep in the Spike gene has driven SARS-CoV-2 human adaptation. Cell. 2021;184(17):4392-4400.e4. pmid:34289344 - 107.
Smith JM, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23(1):23–35. pmid:4407212 - 108.
Levine AJ, Jenkins NA, Copeland NG. The roles of initiating truncal mutations in human cancers: the order of mutations and tumor cell type matters. Cancer Cell. 2019;35(1):10–5. pmid:30645969 - 109.
Hoemberger M, Pitsawong W, Kern D. Cumulative mechanism of several major imatinib-resistant mutations in Abl kinase. Proc Natl Acad Sci U S A. 2020;117(32):19221–7. pmid:32719139 - 110.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. pmid:21376230 - 111.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. pmid:35022204 - 112.
Noble D. Neo-Darwinism, the modern synthesis and selfish genes: are they of use in physiology?. J Physiol. 2011;589(Pt 5):1007–15. pmid:21135048 - 113.
Kimura M, Crow JF. The number of alleles that can be maintained in a finite population. Genetics. 1964;49(4):725–38. pmid:14156929 - 114.
Cannataro VL, Townsend JP. Neutral theory and the somatic evolution of cancer. Mol Biol Evol. 2018;35(6):1308–15. pmid:29684198 - 115.
Bozic I, Paterson C, Waclaw B. On measuring selection in cancer from subclonal mutation frequencies. PLoS Comput Biol. 2019;15(9):e1007368. pmid:31557163 - 116.
Madan E, Pelham CJ, Nagane M, Parker TM, Canas-Marques R, Fazio K, et al. Flower isoforms promote competitive growth in cancer. Nature. 2019;572(7768):260-4. pmid:31341286. - 117.
Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514(7520):54–8. pmid:25079331 - 118.
West J, Newton PK. Cellular interactions constrain tumor growth. Proc Natl Acad Sci U S A. 2019;116(6):1918–23. pmid:30674661 - 119.
Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463(7280):545–8. pmid:20072127 - 120.
Gould SJ. Is a new and general theory of evolution emerging?. Paleobiology. 1980;6(1):119–30. - 121.
Gould SJ. Evolution: the pleasures of pluralism. The New York Review of Books,. 1997 June 26. - 122.
Lewontin RC. The triple helix: gene, organism, and environment. Reprint edition (2001) ed. Cambridge: Harvard University Press; 2000. - 123.
Laland K, Uller T, Feldman M, Sterelny K, Müller GB, Moczek A, et al. Does evolutionary theory need a rethink?. Nature. 2014;514(7521):161–4. pmid:25297418 - 124.
Vendramin R, Litchfield K, Swanton C. Cancer evolution: Darwin and beyond. EMBO J. 2021;40(18):e108389. pmid:34459009 - 125.
Eldredge N, Gould SJ. On punctuated equilibria. Science. 1997;276(5311):338–41. pmid:9139351 - 126.
Gould SJ. Tempo and mode in the macroevolutionary reconstruction of Darwinism. Proc Natl Acad Sci U S A. 1994;91(15):6764–71. pmid:8041695 - 127.
Leigh EG Jr. The group selection controversy. J Evol Biol. 2010;23(1):6–19. pmid:20002254 - 128.
Borges RM. Co-niche construction between hosts and symbionts: ideas and evidence. J Genet. 2017;96(3):483–9. pmid:28761011 - 129.
Webster G, Goodwin BC. A structuralist approach to morphology. Riv Biol. 1999;92(3):495–8. pmid:10765682 - 130.
Crispo E. The Baldwin effect and genetic assimilation: revisiting two mechanisms of evolutionary change mediated by phenotypic plasticity. Evolution. 2007;61(11):2469–79. pmid:17714500 - 131.
West-Eberhard MJ. Developmental plasticity and the origin of species differences. Proc Natl Acad Sci U S A. 2005;102 Suppl 1(Suppl 1):6543–9. pmid:15851679 - 132.
Waddington C. Genetic assimilation of the bithorax phenotype. Evolution. 1956;10. - 133.
Morgan TJH, Suchow JW, Griffiths TL. What the Baldwin Effect affects depends on the nature of plasticity. Cognition. 2020;197:104165. pmid:31958668 - 134.
Waddington CH. Canalization of development and the inheritance of acquired characters. Nature. 1942;150(3811):563–5. - 135.
Schwander T, Leimar O. Genes as leaders and followers in evolution. Trends Ecol Evol. 2011;26(3):143–51. pmid:21257223 - 136.
Lazebnik Y. Are scientists a workforce?—Or, how Dr. Frankenstein made biomedical research sick: a proposed plan to rescue US biomedical research from its current “malaise” will not be effective as it misdiagnoses the root cause of the disease. EMBO Rep. 2015;16(12):1592–600. pmid:26566662 - 137.
Demicheli R, Hrushesky WJM. Reimagining cancer: moving from the cellular to the tissue level. Cancer Res. 2023;83(2):173–80. pmid:36264185 - 138.
Soto AM, Longo G, Miquel P-A, Montevil M, Mossio M, Perret N, et al. Toward a theory of organisms: three founding principles in search of a useful integration. Prog Biophys Mol Biol. 2016;122(1):77–82. pmid:27498204 - 139.
Geman D, Geman S. Opinion: science in the age of selfies. Proc Natl Acad Sci U S A. 2016;113(34):9384–7. pmid:27555443 - 140.
Ball P. How life works: a user’s guide to the new biology: University of Chicago Press; 2023. - 141.
Strohman RC. The coming Kuhnian revolution in biology. Nat Biotechnol. 1997;15(3):194–200. pmid:9062910 - 142.
Baker SG. A cancer theory kerfuffle can lead to new lines of research. J Natl Cancer Inst. 2014;107(2):dju405. pmid:25528755 - 143.
Blagosklonny MV. Molecular theory of cancer. Cancer Biol Ther. 2005;4(6):621–7. pmid:15970666 - 144.
Satgé D. Analysis of somatic mutations in cancer tissues challenges the somatic mutation theory of cancer. Encyclopedia of Life Sciences. 2013. - 145.
Sonnenschein C, Soto AM. Over a century of cancer research: inconvenient truths and promising leads. PLoS Biol. 2020;18(4):e3000670. pmid:32236102 - 146.
Pierce GB. On the boundary between development and neoplasia. An interview with Professor G. Barry Pierce. Interview by Juan Arechaga. Int J Dev Biol. 1993;37(1):5–16. pmid:8507570 - 147.
Pierce GB, Aguilar D, Hood G, Wells RS. Trophectoderm in control of murine embryonal carcinoma. Cancer Res. 1984;44(9):3987–96. pmid:6744314 - 148.
Sell S, Pierce GB. Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab Invest. 1994;70(1):6–22. pmid:8302019 - 149.
Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci U S A. 1975;72(9):3585–9. pmid:1059147 - 150.
Smithers DW. Maturation in human tumours. Lancet. 1969;2(7627):949–52. pmid:4186609 - 151.
Ambros IM, Zellner A, Roald B, Amann G, Ladenstein R, Printz D, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med. 1996;334(23):1505–11. pmid:8618605 - 152.
Fathi AT, Stein EM, DiNardo CD, Levis MJ, Montesinos P, de Botton S. Differentiation syndrome with lower-intensity treatments for acute myeloid leukemia. Am J Hematol. 2021;96(6):735–46. pmid:33625753 - 153.
McCullough KD, Coleman WB, Ricketts SL, Wilson JW, Smith GJ, Grisham JW. Plasticity of the neoplastic phenotype in vivo is regulated by epigenetic factors. Proc Natl Acad Sci U S A. 1998;95(26):15333–8. pmid:9860969 - 154.
Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117(Pt 8):1495–502. pmid:14996910 - 155.
Kenny PA, Bissell MJ. Tumor reversion: correction of malignant behavior by microenvironmental cues. Int J Cancer. 2003;107(5):688–95. pmid:14566816 - 156.
Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231–45. pmid:9105051 - 157.
Hendrix MJC, Seftor EA, Seftor REB, Kasemeier-Kulesa J, Kulesa PM, Postovit L-M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7(4):246–55. pmid:17384580 - 158.
Huang S. The logic of cancer treatment: why it is so hard to cure cancertreatment-induced progression, hyper-progression and the Nietzsche effect. In: Strauss B, Bertolaso M, Ernberg I, Bissell MJ, editors. Rethinking cancer: a new paradigm for the post-genomics era Vienna series in theoretical biology. Cambridge, MA: MIT Press; 2021. p. 456. - 159.
Stevens LC. The development of transplantable teratocarcinomas from intratesticular grafts of pre- and postimplantation mouse embryos. Dev Biol. 1970;21(3):364–82. pmid:5436899 - 160.
Noble D. The music of life: biology beyond gene. Oxford: Oxford University Press;2006. - 161.
Kupiec JJ. A probabilist theory for cell differentiation, embryonic mortality and DNA C-value paradox. Speculation Sci Technol. 1983;6(5):471–8. - 162.
Huang S. The molecular and mathematical basis of Waddington’s epigenetic landscape: a framework for post-Darwinian biology? Bioessays. 2012;34(2):149–57. pmid:22102361 - 163.
MONOD J, JACOB F. Teleonomic mechanisms in cellular metabolism, growth, and differentiation. Cold Spring Harb Symp Quant Biol. 1961;26:389–401. pmid:14475415 - 164.
Kauffman S. Homeostasis and differentiation in random genetic control networks. Nature. 1969;224(5215):177–8. pmid:5343519 - 165.
Kauffman SA. The origins of order. New York: Oxford University Press; 1993. - 166.
Huang S. Reprogramming cell fates: reconciling rarity with robustness. Bioessays. 2009;31(5):546–60. pmid:19319911 - 167.
Huang S. Towards a unification of the 2 meanings of “epigenetics”. PLoS Biol. 2022;20(12):e3001944. pmid:36574409 - 168.
Davidson EH, Erwin DH. Gene regulatory networks and the evolution of animal body plans. Science. 2006;311(5762):796–800. pmid:16469913 - 169.
Arias AM, Hayward P. Filtering transcriptional noise during development: concepts and mechanisms. Nat Rev Genet. 2006;7(1):34–44. pmid:16369570 - 170.
Blake WJ, Balázsi G, Kohanski MA, Isaacs FJ, Murphy KF, Kuang Y, et al. Phenotypic consequences of promoter-mediated transcriptional noise. Mol Cell. 2006;24(6):853–65. pmid:17189188 - 171.
Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136(23):3853–62. pmid:19906852 - 172.
Zhou JX, Aliyu MDS, Aurell E, Huang S. Quasi-potential landscape in complex multi-stable systems. J R Soc Interface. 2012;9(77):3539–53. pmid:22933187 - 173.
Sáez M, Briscoe J, Rand DA. Dynamical landscapes of cell fate decisions. Interface Focus. 2022;12(4):20220002. pmid:35860004 - 174.
Goldbeter A. Dissipative structures in biological systems: bistability, oscillations, spatial patterns and waves. Philos Trans A Math Phys Eng Sci. 2018;376(2124):20170376. pmid:29891498 - 175.
Wang J, Xu L, Wang E. Potential landscape and flux framework of nonequilibrium networks: robustness, dissipation, and coherence of biochemical oscillations. Proc Natl Acad Sci U S A. 2008;105(34):12271–6. pmid:18719111 - 176.
Huang S. Genetic and non-genetic instability in tumor progression: link between the fitness landscape and the epigenetic landscape of cancer cells. Cancer Metastasis Rev. 2013;32(3–4):423–48. pmid:23640024 - 177.
Kauffman S. Differentiation of malignant to benign cells. J Theor Biol. 1971;31(3):429–51. pmid:5556142 - 178.
Huang S, Ernberg I, Kauffman S. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin Cell Dev Biol. 2009;20(7):869–76. pmid:19595782 - 179.
Raff MC. Social controls on cell survival and cell death. Nature. 1992;356(6368):397–400. pmid:1557121 - 180.
Vainshelbaum NM, Giuliani A, Salmina K, Pjanova D, Erenpreisa J. The transcriptome and proteome networks of malignant tumours reveal atavistic attractors of polyploidy-related asexual reproduction. Int J Mol Sci. 2022;23(23):14930. pmid:36499258 - 181.
Vincent M. Cancer: a de-repression of a default survival program common to all cells?: a life-history perspective on the nature of cancer. Bioessays. 2012;34(1):72–82. pmid:22105565 - 182.
Takahashi H. Memoirs of a neuropathologist who was unfit to be a clinician. Free Neuropathol. 2023;4:4–7. pmid:37347035 - 183.
Soto AM, Sonnenschein C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory. Bioessays. 2011;33(5):332–40. pmid:21503935 - 184.
Sharma V, Letson J, Furuta S. Fibrous stroma: driver and passenger in cancer development. Sci Signal. 2022;15(724):eabg3449. pmid:35258999 - 185.
Jasencakova Z, Groth A. Replication stress, a source of epigenetic aberrations in cancer?. Bioessays. 2010;32(10):847–55. pmid:20726011 - 186.
Saxena S, Zou L. Hallmarks of DNA replication stress. Mol Cell. 2022;82(12):2298–314. pmid:35714587 - 187.
Telerman A, Amson R. The molecular programme of tumour reversion: the steps beyond malignant transformation. Nat Rev Cancer. 2009;9(3):206–16. pmid:19180095 - 188.
Cunha GR, Hayashi N, Wong YC. Regulation of differentiation and growth of normal adult and neoplastic epithelial by inductive mesenchyme. In: Isaacs JT, editor. Prostate cancer: cell and molecular mechanisms in diagnosis and treatment. Cold Spring Harbor,NY: Cold Spring Harbor Laboratory Press,; 1991. p. 73–90. - 189.
Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol. 2004;51(1):1–28. pmid:15207251 - 190.
de Thé H. Differentiation therapy revisited. Nat Rev Cancer. 2018;18(2):117–27. pmid:29192213 - 191.
Chang WY. Complete spontaneous regression of cancer: four case reports, review of literature, and discussion of possible mechanisms involved. Hawaii Med J. 2000;59(10):379–87. pmid:11789163 - 192.
Gilbert SF. Commentary: “The epigenotype” by C.H. Waddington. Int J Epidemiol. 2012;41(1):20–3. pmid:22253306 - 193.
Morinishi L, Kochanowski K, Levine RL, Wu LF, Altschuler SJ. Loss of TET2 affects proliferation and drug sensitivity through altered dynamics of cell-state transitions. Cell Syst. 2020;11(1):86–94.e5. pmid:32619551 - 194.
Sharma S, Rani H, Mahesh Y, Jolly MK, Dixit J, Mahadevan V. Loss of p53 epigenetically modulates epithelial to mesenchymal transition in colorectal cancer. Transl Oncol. 2024;43:101848. pmid:38412660 - 195.
Aldana M, Balleza E, Kauffman S, Resendiz O. Robustness and evolvability in genetic regulatory networks. J Theor Biol. 2007;245(3):433–48. pmid:17188715 - 196.
Balleza E, Alvarez-Buylla ER, Chaos A, Kauffman S, Shmulevich I, Aldana M. Critical dynamics in genetic regulatory networks: examples from four kingdoms. PLoS One. 2008;3(6):e2456. pmid:18560561 - 197.
Ye CJ, Regan S, Liu G, Alemara S, Heng HH. Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol Cytogenet. 2018;11:31. pmid:29760781 - 198.
Liu J, Niu N, Li X, Zhang X, Sood AK. The life cycle of polyploid giant cancer cells and dormancy in cancer: opportunities for novel therapeutic interventions. Semin Cancer Biol. 2022;81:132–44. pmid:34670140 - 199.
Erenpreisa J, Cragg MS. Cancer: a matter of life cycle? Cell Biol Int. 2007;31(12):1507–10. pmid:17936649 - 200.
Liu J. The “life code”: a theory that unifies the human life cycle and the origin of human tumors. Semin Cancer Biol. 2020;60:380–97. pmid:31521747 - 201.
Gurjao C, Tsukrov D, Imakaev M, Luquette LJ, Mirny LA. Is tumor mutational burden predictive of response to immunotherapy? eLife Sciences Publications; 2024. - 202.
Prehn RT. Cancers beget mutations versus mutations beget cancers. Cancer Res. 1994;54(20):5296–300. pmid:7923156 - 203.
Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. The DNA damage response induces IFN. J Immunol. 2011;187(10):5336–45. pmid:22013119 - 204.
Tong J, Song J, Zhang W, Zhai J, Guan Q, Wang H, et al. When DNA-damage responses meet innate and adaptive immunity. Cell Mol Life Sci. 2024;81(1):185. pmid:38630271 - 205.
Smithers DW. An attack on cytologism. Lancet. 1962;1(7228):493–9. pmid:13914465 - 206.
Tlsty TD, Hein PW. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev. 2001;11(1):54–9. pmid:11163151 - 207.
Virchow RLK. Cellular pathology. 1859 special ed., 204–207 London, UK: John Churchill 1978. - 208.
Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–11. pmid:10519415 - 209.
Tlsty TD, Gascard P. Stromal directives can control cancer. Science. 2019;365(6449):122–3. pmid:31296756 - 210.
Zarbl H, Sukumar S, Arthur AV, Martin-Zanca D, Barbacid M. Direct mutagenesis of Ha-ras-1 oncogenes by N-nitroso-N-methylurea during initiation of mammary carcinogenesis in rats. Nature. 1985;315(6018):382–5. pmid:3923365 - 211.
Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? Bioessays. 2004;26(10):1097–107. pmid:15382143 - 212.
Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–50. pmid:18039110 - 213.
Gilbert SF, Opitz JM, Raff RA. Resynthesizing evolutionary and developmental biology. Dev Biol. 1996;173(2):357–72. pmid:8605997 - 214.
Rawles ME. Tissue interactions in scale and feather development as studied in dermal-epidermal recombinations. J Embryol Exp Morphol. 1963;11:765–89. pmid:14081994 - 215.
Sakakura T, Suzuki Y, Shiurba R. Mammary stroma in development and carcinogenesis. J Mammary Gland Biol Neoplasia. 2013;18(2):189–97. pmid:23604977 - 216.
Cha RS, Guerra L, Thilly WG, Zarbl H. Ha-ras-1 oncogene mutations in mammary epithelial cells do not contribute to initiation of spontaneous mammary tumorigenesis in rats. Carcinogenesis. 1996;17(11):2519–24. pmid:8968072 - 217.
Barcellos-Hoff MH, Ravani SA. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60(5):1254–60. pmid:10728684 - 218.
Hayward SW, Wang Y, Cao M, Hom YK, Zhang B, Grossfeld GD, et al. Malignant transformation in a nontumorigenic human prostatic epithelial cell line. Cancer Res. 2001;61(22):8135–42. pmid:11719442 - 219.
Barclay WW, Woodruff RD, Hall MC, Cramer SD. A system for studying epithelial-stromal interactions reveals distinct inductive abilities of stromal cells from benign prostatic hyperplasia and prostate cancer. Endocrinology. 2005;146(1):13–8. pmid:15471963 - 220.
Lee S-H, Contreras Panta EW, Gibbs D, Won Y, Min J, Zhang C, et al. Apposition of fibroblasts with metaplastic gastric cells promotes dysplastic transition. Gastroenterology. 2023;165(2):374–90. pmid:37196797 - 221.
Maffini MV, Calabro JM, Soto AM, Sonnenschein C. Stromal regulation of neoplastic development: age-dependent normalization of neoplastic mammary cells by mammary stroma. Am J Pathol. 2005;167(5):1405–10. pmid:16251424 - 222.
Kasemeier-Kulesa JC, Teddy JM, Postovit L-M, Seftor EA, Seftor REB, Hendrix MJC, et al. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev Dyn. 2008;237(10):2657–66. pmid:18629870 - 223.
McCullough KD, Coleman WB, Smith GJ, Grisham JW. Age-dependent induction of hepatic tumor regression by the tissue microenvironment after transplantation of neoplastically transformed rat liver epithelial cells into the liver. Cancer Res. 1997;57(9):1807–13. pmid:9135026 - 224.
Bussard KM, Boulanger CA, Booth BW, Bruno RD, Smith GH. Reprogramming human cancer cells in the mouse mammary gland. Cancer Res. 2010;70(15):6336–43. pmid:20647316 - 225.
Bussard KM, Smith GH. Human breast cancer cells are redirected to mammary epithelial cells upon interaction with the regenerating mammary gland microenvironment in-vivo. PLoS One. 2012;7(11):e49221. pmid:23155468 - 226.
Bischof AG, Yüksel D, Mammoto T, Mammoto A, Krause S, Ingber DE. Breast cancer normalization induced by embryonic mesenchyme is mediated by extracellular matrix biglycan. Integr Biol (Camb). 2013;5(8):1045–56. pmid:23817524 - 227.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34. pmid:24856586 - 228.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–47. pmid:24856585 - 229.
Su H, Yang F, Fu R, Trinh B, Sun N, Liu J, et al. Collagenolysis-dependent DDR1 signalling dictates pancreatic cancer outcome. Nature. 2022;610(7931):366–72. pmid:36198801 - 230.
Bischoff F, Bryson G. Carcinogenesis through solid state surfaces. Prog Exp Tumor Res. 1964;5:85–133. pmid:14317768 - 231.
Clark WH Jr. The nature of cancer: morphogenesis and progressive (self)-disorganization in neoplastic development and progression. Acta Oncol. 1995;34(1):3–21. pmid:7865232 - 232.
Huang S, Ingber DE. Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005;8(3):175–6. pmid:16169461 - 233.
Ingber DE. Cancer as a disease of epithelial-mesenchymal interactions and extracellular matrix regulation. Differentiation. 2002;70(9–10):547–60. pmid:12492496 - 234.
Northcott JM, Dean IS, Mouw JK, Weaver VM. Feeling stress: the mechanics of cancer progression and aggression. Front Cell Dev Biol. 2018;6:17. pmid:29541636 - 235.
Montévil M, Mossio M. Biological organisation as closure of constraints. J Theor Biol. 2015;372:179–91. pmid:25752259 - 236.
Soto AM, Sonnenschein C. The cancer puzzle: welcome to organicism. Prog Biophys Mol Biol. 2021;165:114–9. pmid:34271028 - 237.
Peltomäki P. Mutations and epimutations in the origin of cancer. Exp Cell Res. 2012;318(4):299–310. pmid:22182599 - 238.
BERENBLUM I, SHUBIK P. A new, quantitative, approach to the study of the stages of chemical carcinogenesis in the mouse’s skin. Br J Cancer. 1947;1(4):383–91. pmid:18906316 - 239.
Balmain A. The critical roles of somatic mutations and environmental tumor-promoting agents in cancer risk. Nat Genet. 2020;52(11):1139–43. pmid:33106632 - 240.
Iversen OH. The reverse experiment in two-stage skin carcinogenesis. It cannot be genuinely performed, but when approximated, it is not innocuous. APMIS Suppl. 1993;34:1–96. pmid:8251199 - 241.
Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284–99. pmid:26972587 - 242.
Huang S, Ingber DE. The structural and mechanical complexity of cell-growth control. Nat Cell Biol. 1999;1(5):E131-8. pmid:10559956 - 243.
Huang S, Ingber DE. Shape-dependent control of cell growth, differentiation, and apoptosis: switching between attractors in cell regulatory networks. Exp Cell Res. 2000;261(1):91–103. pmid:11082279 - 244.
Lakatos I. Falsification and the methodology of scientific research programmes. In: Musgrave A, Lakatos I, editors. Criticism and the growth of knowledge: proceedings of the international colloquium in the philosophy of science, London, 1965. 4. Cambridge: Cambridge University Press; 1970. p. 91–196. - 245.
Quine WVO. Two dogmas of empiricism. Philos Rev. 1951;60(1):20–43. - 246.
Feynman R. The character of physical law, with new foreword: MIT Press; 2017. - 247.
Darwin Correspondence Project Ln, To Henry Fawcett 18 September [1861] 1861 [3 February 2017]. Available from: http://www.darwinproject.ac.uk/DCP-LETT-3257. - 248.
Kozlov M. “Disruptive” science has declined—and no one knows why. Nature. 2023;613(7943):225. pmid:36599999
ADVERTISEMENT:
Halo, para pengemar slot! pernahkah mendengar istilah “raja slot? Kalau belum, siap-siap jatuh cinta dengan konsep ini. slot gaco adalah mesin slots yang selalu kasih kemenangan. Yup, mesin-mesin ini bisa disebut sebagai jagoannya buat bawa come back hasil. tapi, cemana sih caranya nemuin slot gacor yang tepat? Santai Bro and Sis, kita bahas tenang saja di sini
Gaming terpercaya saat ini hanya satu berada Indonesia hanya di pasti menyediakan return on Investment tertinggi
Daftar dengan di :
Informasi mengenai KING SLOT, Segera Daftar Bersama king selot terbaik dan terpercaya no satu di Indonesia. Boleh mendaftar melalui sini king slot serta memberikan hasil kembali yang paling tinggi saat sekarang ini hanyalah KING SLOT atau Raja slot paling gacor, gilak dan gaco saat sekarang di Indonesia melalui program return tinggi di kingselot serta pg king slot
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
situs slot terbaru
situs slot terbaru permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
slot terbaru
slot terbaru permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
suara88 permainan paling top dan garansi imbal balik hasil besar bersama suara88.biz
sumo7777 permainan paling top dan garansi imbal balik hasil besar bersama sumo7777.com
supermoney888 permainan paling top dan garansi imbal balik hasil besar bersama supermoney888.biz
teratai88 permainan paling top dan garansi imbal balik hasil besar bersama teratai88.biz
thor88 permainan paling top dan garansi imbal balik hasil besar bersama thor88.biz
togelhk88 permainan paling top dan garansi imbal balik hasil besar bersama togelhk88.net
topjitu88 permainan paling top dan garansi imbal balik hasil besar bersama topjitu88.net
totosloto88 permainan paling top dan garansi imbal balik hasil besar bersama totosloto88.com
trisula888 permainan paling top dan garansi imbal balik hasil besar bersama trisula888.biz
udangbet88 permainan paling top dan garansi imbal balik hasil besar bersama udangbet88.net
via88 permainan paling top dan garansi imbal balik hasil besar bersama via88.biz
virusjp88 permainan paling top dan garansi imbal balik hasil besar bersama virusjp88.net
warga888 permainan paling top dan garansi imbal balik hasil besar bersama warga888.biz
waw88 permainan paling top dan garansi imbal balik hasil besar bersama waw88.biz
winjitu88 permainan paling top dan garansi imbal balik hasil besar bersama winjitu88.net
wisdom88 permainan paling top dan garansi imbal balik hasil besar bersama wisdom88.biz
wnitogel88 permainan paling top dan garansi imbal balik hasil besar bersama wnitogel88.com
yoyo888 permainan paling top dan garansi imbal balik hasil besar bersama yoyo888.biz
validtoto88 permainan paling top dan garansi imbal balik hasil besar bersama validtoto88.com
sule999 permainan paling top dan garansi imbal balik hasil besar bersama sule999.com
sule88 permainan paling top dan garansi imbal balik hasil besar bersama sule88.org
ss888bet permainan paling top dan garansi imbal balik hasil besar bersama ss888bet.com
sia77 permainan paling top dan garansi imbal balik hasil besar bersama sia77.info
seluang88 permainan paling top dan garansi imbal balik hasil besar bersama seluang88.com
satu88 permainan paling top dan garansi imbal balik hasil besar bersama satu88.biz
satu777 permainan paling top dan garansi imbal balik hasil besar bersama satu777.asia
rp88 permainan paling top dan garansi imbal balik hasil besar bersama rp88.biz
rp88 permainan paling top dan garansi imbal balik hasil besar bersama rp88.asia
rp88 permainan paling top dan garansi imbal balik hasil besar bersama rp77.live
qiuqiu88 permainan paling top dan garansi imbal balik hasil besar bersama qiuqiu88.biz
pt88 permainan paling top dan garansi imbal balik hasil besar bersama pt88.org
pt77 permainan paling top dan garansi imbal balik hasil besar bersama pt77.info
produk88 permainan paling top dan garansi imbal balik hasil besar bersama produk88.asia
mt88 permainan paling top dan garansi imbal balik hasil besar bersama mt88.org
mt77 permainan paling top dan garansi imbal balik hasil besar bersama mt77.biz
menang66 permainan paling top dan garansi imbal balik hasil besar bersama menang66.biz
latobet888 permainan paling top dan garansi imbal balik hasil besar bersama latobet888.org
kedai96 permainan paling top dan garansi imbal balik hasil besar bersama kedai96.org
kedai188 permainan paling top dan garansi imbal balik hasil besar bersama kedai188.biz
ids88 permainan paling top dan garansi imbal balik hasil besar bersama ids88.biz
hp88 permainan paling top dan garansi imbal balik hasil besar bersama hp88.org
hp77 permainan paling top dan garansi imbal balik hasil besar bersama hp77.org
gm88 permainan paling top dan garansi imbal balik hasil besar bersama gm88.asia
gm77 permainan paling top dan garansi imbal balik hasil besar bersama gm77.net
final888 permainan paling top dan garansi imbal balik hasil besar bersama final888.org
duit88 permainan paling top dan garansi imbal balik hasil besar bersama duit88.asia
duit168 permainan paling top dan garansi imbal balik hasil besar bersama duit168.biz
divisi88 permainan paling top dan garansi imbal balik hasil besar bersama divisi88.org
dewi500 permainan paling top dan garansi imbal balik hasil besar bersama dewi500.biz
devil88 permainan paling top dan garansi imbal balik hasil besar bersama devil88.info
cuputoto88 permainan paling top dan garansi imbal balik hasil besar bersama cuputoto88.com
cukongbet88 permainan paling top dan garansi imbal balik hasil besar bersama cukongbet88.asia
bom888 permainan paling top dan garansi imbal balik hasil besar bersama bom888.biz
bintaro888 permainan paling top dan garansi imbal balik hasil besar bersama bintaro888.info
askasino88 permainan paling top dan garansi imbal balik hasil besar bersama askasino88.org
999aset permainan paling top dan garansi imbal balik hasil besar bersama 999aset.com
afb77 permainan paling top dan garansi imbal balik hasil besar bersama afb77.biz
aset99 permainan paling top dan garansi imbal balik hasil besar bersama aset99.biz
bendera77 permainan paling top dan garansi imbal balik hasil besar bersama bendera77.biz
bendera888 permainan paling top dan garansi imbal balik hasil besar bersama bendera888.com
coco88 permainan paling top dan garansi imbal balik hasil besar bersama coco88.org
cuma77 permainan paling top dan garansi imbal balik hasil besar bersama cuma77.biz
cuma88 permainan paling top dan garansi imbal balik hasil besar bersama cuma88.org
dwv88 permainan paling top dan garansi imbal balik hasil besar bersama dwv88.org
fafajp88 permainan paling top dan garansi imbal balik hasil besar bersama fafajp88.com
gemar88 permainan paling top dan garansi imbal balik hasil besar bersama gemar88.biz
gocap88 permainan paling top dan garansi imbal balik hasil besar bersama gocap88.info
gocaptoto permainan paling top dan garansi imbal balik hasil besar bersama gocaptoto.asia
hakabet88 permainan paling top dan garansi imbal balik hasil besar bersama hakabet88.com
hwtoto88 permainan paling top dan garansi imbal balik hasil besar bersama hwtoto88.org
ina77 permainan paling top dan garansi imbal balik hasil besar bersama ina77.biz
ina88 permainan paling top dan garansi imbal balik hasil besar bersama ina88.info
jingga8888 permainan paling top dan garansi imbal balik hasil besar bersama jingga8888.com
juragan777 permainan paling top dan garansi imbal balik hasil besar bersama juragan777.asia
kastil77 permainan paling top dan garansi imbal balik hasil besar bersama kastil77.info
kebo888 permainan paling top dan garansi imbal balik hasil besar bersama kebo888.biz
kkwin77 permainan paling top dan garansi imbal balik hasil besar bersama kkwin77.com
kokoslot88 permainan paling top dan garansi imbal balik hasil besar bersama kokoslot88.asia
luckydf88 permainan paling top dan garansi imbal balik hasil besar bersama luckydf88.org
microstar888 permainan paling top dan garansi imbal balik hasil besar bersama microstar888.biz
monperatoto88 permainan paling top dan garansi imbal balik hasil besar bersama monperatoto88.com
mpo1122 permainan paling top dan garansi imbal balik hasil besar bersama mpo1122.biz
mpo122 permainan paling top dan garansi imbal balik hasil besar bersama mpo122.biz
mpopelangi88 permainan paling top dan garansi imbal balik hasil besar bersama mpopelangi88.com
pamanslot88 permainan paling top dan garansi imbal balik hasil besar bersama pamanslot88.biz
panel88 permainan paling top dan garansi imbal balik hasil besar bersama panel88.org
paragon77 permainan paling top dan garansi imbal balik hasil besar bersama paragon77.biz
paragon888 permainan paling top dan garansi imbal balik hasil besar bersama paragon888.info
pion77 permainan paling top dan garansi imbal balik hasil besar bersama pion77.biz
prada88 permainan paling top dan garansi imbal balik hasil besar bersama prada88.asia
prada888 permainan paling top dan garansi imbal balik hasil besar bersama prada888.com
qqslot88slot permainan paling top dan garansi imbal balik hasil besar bersama qqslot88slot.com
rejekibet88 permainan paling top dan garansi imbal balik hasil besar bersama rejekibet88.com
rezekibet88 permainan paling top dan garansi imbal balik hasil besar bersama rezekibet88.org
sensa77 permainan paling top dan garansi imbal balik hasil besar bersama sensa77.biz
sensa888 permainan paling top dan garansi imbal balik hasil besar bersama sensa888.biz
singajp88 permainan paling top dan garansi imbal balik hasil besar bersama singajp88.com
sr77 permainan paling top dan garansi imbal balik hasil besar bersama sr77.org
sr88 permainan paling top dan garansi imbal balik hasil besar bersama sr88.org
surya77 permainan paling top dan garansi imbal balik hasil besar bersama surya77.biz
surya88 permainan paling top dan garansi imbal balik hasil besar bersama surya88.asia
tajir77 permainan paling top dan garansi imbal balik hasil besar bersama tajir77.info
tajir88 permainan paling top dan garansi imbal balik hasil besar bersama tajir88.biz
toto122 permainan paling top dan garansi imbal balik hasil besar bersama toto122.com
toto123 permainan paling top dan garansi imbal balik hasil besar bersama toto123.biz
uangvip88 permainan paling top dan garansi imbal balik hasil besar bersama uangvip88.com
wajik77 permainan paling top dan garansi imbal balik hasil besar bersama wajik77.asia
777neko permainan paling top dan garansi imbal balik hasil besar bersama 777neko.org
88judi permainan paling top dan garansi imbal balik hasil besar bersama 88judi.net
99judi permainan paling top dan garansi imbal balik hasil besar bersama 99judi.org
abcslot88 permainan paling top dan garansi imbal balik hasil besar bersama abcslot88.asia



