









Abstract
Bacterial noncoding RNAs fulfill a variety of cellular functions as catalysts, as scaffolds in protein complexes or as regulators of gene expression. They often exhibit complex tertiary structures that are a key determinant of their biochemical function. Here, we characterize the structured “raiA motif” RNA from Clostridioides difficile, which is conserved in more than 2,500 bacterial species from the phyla Bacillota and Actinomycetota. We show that its transcript abundance and stability in exponentially growing bacteria rivals that of ribosomal RNAs. Deletion of the “raiA motif” RNA is associated with delayed transition into stationary phase, and changes in stationary phase pathways such as spore formation, hence we rename it ModT (modulator of transition phase). Mechanistically, we show that ModT-mediated changes in cellular cyclic di-GMP levels are linked to the pronounced sporulation defect in the modT mutant. Importantly, we show that expression profiles and isoform patterns of ModT are conserved in Clostridium perfringens and Paeniclostridium sordellii, and that these orthologs can functionally complement ModT in C. difficile. Chemical structure probing of ModT in vivo reveals dynamic refolding and provides initial evidence for a potential association of ModT with proteins. In summary, our findings indicate that ModT fulfills a conserved role in regulating growth transitions in bacteria and provide a crucial step towards delineating its molecular mechanism.
Citation: Lenče T, Sulzer J, Andress K, Gribling-Burrer A-S, Lamm-Schmidt V, Barquist L, et al. (2024) The conserved noncoding RNA ModT coordinates growth and virulence in Clostridioides difficile. PLoS Biol 22(12):
e3002948.
https://doi.org/10.1371/journal.pbio.3002948
Academic Editor: Matthew K. Waldor, Brigham and Women’s Hospital, UNITED STATES OF AMERICA
Received: September 18, 2024; Accepted: November 22, 2024; Published: December 13, 2024
Copyright: © 2024 Lenče et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Data Availability: All RNA-sequencing data, either re-used or generated in this study, are available at the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo) under the accession numbers GSE165672 (Grad-seq, S1 Dataset), GSE273118 (RNA-seq, S2 Dataset) and GSE273119 (DMS-MaP structure probing). All data underlying figures within this manuscript can be found in S5 Dataset. All original and uncropped minimally adjusted images of RNA gels and northern blots shown within the manuscript can be found in S1 Raw images.
Funding: J.S. was supported by the German Research Foundation (https://www.dfg.de/en, grant no. FA 1113/2-1). R.P.S was supported by the Helmholtz Association (https://www.helmholtz.de/en/, grant no. VH-NG-1347). T.L. was supported by the Bavarian State Ministry for Science and the Arts through the research network bayresq.net (https://bayresq.net/en/startseite-en/). T.L. was also supported through the Joachim Herz Foundation (https://www.joachim-herz-stiftung.de/en/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have declared that no competing interests exist.
Abbreviations:
BHI,
brain heart infusion; DMS-MaP,
dimethyl sulfate mutational profiling; EP,
exponential phase; FDR,
false discovery rate; ILP,
in-line probing; LB,
Luria–Bertani; ncRNA,
noncoding RNA; OD,
optical density; OLE,
Ornate Large Extremophilic; PAS,
Per-Arnt-Sim; PBS,
phosphate-buffered saline; ROC-AUC,
receiver operator characteristic–area under the curve; RPM,
revolutions per minute; TP,
transition phase; TSS,
transcriptional start site; WT,
wild type
Introduction
Noncoding RNAs (ncRNAs) are central regulators of physiology, immunity and virulence in bacteria [1–3]. They act as ribozymes, scaffolds or regulators of gene expression, by providing fundamental housekeeping functions, defense against genetic invaders, and by optimizing gene expression in response to intracellular or environmental cues [4–6]. To serve this broad spectrum of functions, ncRNAs differ drastically in their sizes, structures, and molecular mechanisms.
The largest group of bacterial ncRNAs comprises small regulatory RNAs (sRNAs) that fine-tune gene expression at the posttranscriptional level. They often act in concert with RNA-binding proteins to regulate translation or mRNA stability, mostly through imperfect base pairing to their target mRNAs [7]. sRNAs typically display narrow conservation within a taxonomic species and are rarely found outside of their bacterial genus or family [8]. In comparison, ncRNAs with exceptional sequence or structural conservation are rare, and they fulfill conserved functions in all bacteria in which they are present. For example, tRNAs and rRNAs contribute to key steps of protein synthesis and are found in all forms of life. Other bacterial ncRNA families with deep structure and/or sequence conservation include 6S RNA, SsrA, the RNA of the Signal Recognition Particle or RNaseP. They all accomplish unique biological functions that are conserved across all bacteria [9–14]. Beyond these few well-known ncRNA families, computational screens of intergenic microbial metagenome sequences have revealed a range of motifs with deep primary sequence and secondary structure conservation [15,16]. While many of them are likely to be cis-acting riboswitches, the size, structural complexity, and genomic location of some motifs make them candidates for large structured RNAs with potential catalytic roles, such as ribozymes [17,18]. Therefore, their characterization holds great promise to reveal novel RNA functions. This is exemplified by the Ornate Large Extremophilic (OLE) RNA, which is encoded in many extremophilic gram-positive bacteria and supports the response to diverse types of stressors in Bacillus halodurans, at least in part by forming a membrane-bound, stress-responsive ribonucleoprotein particle with the OapA protein [19,20].
Using RNA sequencing-based annotation of transcript start sites and transcript ends in the spore-forming anaerobe Clostridioides difficile (C. difficile), we recently characterized the ncRNA landscape in this important nosocomial pathogen [21]. This analysis highlighted expression of an RNA, termed “raiA motif” RNA (RF03072), which is a member of a largely uncharacterized RNA family. Based on our results described below, we have renamed this RNA ModT, for modulator of transition phase (TP). ModT can be found in more than 2,500 biologically diverse species, mostly belonging to the phyla Bacillota and Actinomycetota [15,16]. Intriguingly, a recent study in Clostridium acetobutylicum demonstrated that deletion of the modT locus has a negative impact on spore formation and bacterial aggregation [22]. Sporulation is a wide-spread developmental process in bacteria that allows long-term survival under adverse conditions. In C. difficile, which is the leading cause of healthcare-associated infective diarrhea, spore formation is crucial for host transmission and survival of antibiotic treatment in infected patients [23–25]. Hence, understanding the regulatory principles underpinning this process is crucial to devise much needed therapeutic interventions in the future.
However, the presence of ModT in many nonspore-forming bacteria strongly suggests that it regulates a more fundamental process that is conserved across these biologically diverse modT-encoding bacteria. This hypothesis is further supported by the highly conserved and complex tertiary structure of ModT, that is reminiscent of ribozymes. To gain first insights into potential functions of ModT, Soares and colleagues performed a comprehensive analysis of its gene synteny [22]. Since genes that participate in the same cellular process often co-localize in bacterial genomes, it might indicate potential biochemical and biological functions of ModT. The analysis revealed that comFC genes (74%) and raiA genes (66%) occur most commonly up- or downstream of modT, respectively. In C. difficile, however, the downstream gene is encoding a transcription antiterminator of a PTS operon instead of raiA. In addition to differences in neighboring gene functions, there is variability in the orientation and distance of modT to its neighbor genes across bacteria. Therefore, gene synteny does not appear to a good indicator of potential cellular functions of ModT.
Here, we perform an in vivo characterization of ModT in C. difficile. Our study identifies ModT as an abundant and highly stable trans-acting RNA that impacts the transition from the exponential to the stationary growth phase. This regulatory function of ModT has direct impact on the expression of stationary phase pathways such as sporulation and toxin production, via changes in cellular c-di-GMP levels. We further show that C. difficile ModT can be functionally replaced by several distant orthologs with conserved core structural features. Our work provides functional evidence that ModT fulfills a conserved cellular function that is likely enabled by its highly preserved tertiary structure.
Results
ModT transcript abundance and stability resembles housekeeping RNAs
We previously demonstrated expression of ModT in C. difficile through genome-wide RNA sequencing-based mapping of transcriptional start sites (TSS) and transcript ends (annotated CDIF630nc_001 in [21,26]). ModT caught our attention due to its high sequence and secondary structure conservation across 9 taxonomic classes and 2 bacterial phyla [22]. RNA-seq-based transcript annotation revealed that expression of ModT is driven from a single intergenic TSS that is associated with a canonical housekeeping σA (SigA) promoter (Figs 1A and S1A [21]). Transcription of modT gives rise to a long (ModT (L), 260 nt) and a short (ModT(S), 215 nt) isoform that differ in their 3′ ends (Figs 1B and S1B). Again, our previous RNA-seq-based transcript annotation indicates 2 independent transcription termination events at sites that correspond to transcript lengths detected by northern blot (S1A Fig). Interestingly, analysis of existing RNA-seq data [26] shows that ModT is among the 30 most abundant transcripts in the cell, and only surpassed by some, but not all, ribosomal RNAs and tRNAs, as well as 6S RNA and the signal recognition particle RNA (Fig 1C and S1 Dataset).

Fig 1. ModT is a highly abundant and stable noncoding RNA.
(A) Schematic of the modT locus in C. difficile 630. Promoter region including the SigA -35 box and -10 box are shown on top, TSS and TTS are indicated with an arrow or a pin symbol, respectively. In silico secondary structure prediction (RNAfold) of the modT RNA is displayed in pink, with the 3′-end extension of the long isoform highlighted in purple. (B) ModT expression profile in C. difficile 630 across different growth stages. Total RNA was extracted from C. difficile 630 grown in TY medium supplemented with indicated carbon sources at indicated time points (for reference, see growth curves shown in Fig 3B). ModT transcript was detected by northern blot using a radioactively labeled DNA probe (FFO-67) that captured both ModT isoforms, as indicated with an arrow. 5S rRNA served as loading control. (C) Normalized read distribution of all expressed and annotated transcripts (n = 3,472) in C. difficile 630 sorted by transcript abundance [26]. Inset displays the 50 most abundant transcripts, with ModT labeled in pink. The underlying data can be found in S1 Dataset. (D) In vivo RNA half-lives for different noncoding RNA transcripts. C. difficile 630 was grown to mid-exponential phase and treated with Rifampicin. Total RNA was extracted at the indicated time points and analyzed by northern blotting using radioactively labeled DNA probes. The representative of 3 independent experiments is shown. TSS, transcriptional start site; TTS, transcription termination site.
To develop a better understanding of ModT function, we initially performed northern blot analysis to characterize its expression across growth stages and in the presence of different carbon and energy sources (Figs 1B and S1B). We observed prominent expression of both isoforms in TY (Tryptone-Yeast) medium throughout exponential growth, but it decreased to the limit of detection after transition into the stationary phase. In contrast, during growth on monosaccharides, such as glucose or fructose, steady-state levels of ModT(L), but not ModT(S) remained high even in late stationary phase. Since both isoforms are transcribed from the same promoter, we sought to explore whether their different fates in the stationary phase are determined by different RNA half-lives. Therefore, we monitored RNA levels over time, after Rifampicin-mediated inhibition of de novo modT transcription (Figs 1D and S1C). Intriguingly, the long isoform was substantially more stable (t1/2 = >180 min) than the short isoform (t1/2 = 45.94 min). This suggests that the accumulation of the long isoform in the stationary phase is, at least in part, due to a longer half-life relative to the short isoform. Moreover, the ModT(L) half-life was similar to the housekeeping 5S rRNA (t1/2 = >180 min) and exceeded by far that of the canonical Hfq-dependent small RNAs CDIF630nc_008 (t1/2 = 22.95 min) (Figs 1D and S1C). In addition, we also observed a higher GC content of ModT (45.6% for ModT(S) and 42.6% for ModT(L)) compared to the average GC content of the C. difficile genome (29.1%) or previously annotated sRNAs (27.5%) (S1D Fig). In summary, the ModT transcript exhibits several features that are reminiscent of housekeeping RNAs and suggests that this noncoding RNA fulfills a fundamental physiological function.
ModT isoforms adopt identical structures in vitro and in vivo
The function of many RNA elements is determined by their unique structure [27]. Given that both ModT isoforms exhibit very distinct in vivo half-lives, we wanted to explore whether they fold into distinct structures. First, we compared the C. difficile ModT sequence with 3 structure models for ModT, including the original structure predicted for ModT [15,22], the structure available from the Rfam database (RF03072), and a predicted structure optimized with R-scape (Fig 2A–2C). Of note, all 3 models represent the short isoform, likely because sequence conservation is poor in the extended 3′-end [15], and we appended our predicted terminator sequence to them. The recently updated consensus secondary structure model for ModT [22] has 2 pseudoknots (denoted pk1 and pk2) and 8 predicted base-paired regions (denoted P1 through P8), and except for P2, all of them are present in both isoforms of C. difficile ModT (Fig 2A). We also mapped the C. difficile ModT sequence onto the current Rfam structure (Fig 2B) as well as the R-scape optimized structure (Fig 2C) [28,29]. The 3 models show prominent structural differences in the regions comprising P3 and P4, as well as in the residues involved in potential pseudoknot formations. Therefore, we decided to include all of them in our analysis.

Fig 2. DMS probing uncovers structural dynamics of ModT during different growth phases and test conditions.
(A–C) Manually curated secondary structure models of C. difficile 630 ModT based on available Rfam models (RF3072). The stem loop structure of the appended 3′ end is based on predictions by RNAfold. (A) Consensus secondary structure as described in [15,22], (B) current Rfam structure, and (C) the R-scape optimized structure of C. difficile 630 ModT. Black triangles indicate the most prominent scission events determined by In-line probing (see S2A–S2D Fig). Average DMS-reactivity for each residue, derived from in vitro structure probing of ModT(S) and ModT(L), is coded with a color-gradient (filled circles). (D) DMS reactivity profiles of adenine (A) and cytosine (C) nucleotides for residues 19–210 of ModT in following conditions: in vitro, in vivo native (Nat), and in deproteinated cell lysates (Dep). The Nat and Dep samples were analyzed in EP (3 h), TP (9 h), and stationary phase (24 h) of growth. Under “Nat” and “Dep” conditions, L displays reactivity specific for the long isoform, while S represents combined reactivities of the short and the long RNA isoforms. The conditions are sorted according to sample clustering in S2E Fig. The dendrogram has been omitted for clarity. The underlying data can be found in S5 Dataset. (E) Comparisons of DMS reactivity in different conditions or isoforms. The Δ reactivity for each nucleotide position was calculated by subtraction of normalized DMS reactivities obtained for the indicated conditions (see top of each graph). For “in vivo Native → Δ(ModT(L)—ModT(S))” and “ModT(L) → Δ(Deproteinated–Native),” the mean subtracted reactivity and its standard deviation was calculated from all growth stages (3 h, 9 h, and 24 h). For “In vivo Native → Δ(3h – 9h),” the profiles of ModT(L) and ModT(S) were combined to obtain the mean reactivity and standard deviation at these 2 time points. The underlying data can be found in S5 Dataset. EP, exponential phase; TP, transition phase.
In-line probing assays (ILP) on in vitro transcribed ModT(L) and ModT(S) (Figs 2A–2C and S2A–S2D) provided general support for all 3 in silico predicted structure models. More specifically, ILP indicated prominent cleavage events at nucleotides mostly predicted to be unpaired in each model. Notably, ILP of ModT(L) resulted in a cleavage pattern identical to ModT(S), suggesting that the extended 3′-end adopts a fold that does not alter the core structure of ModT(L). However, given that the structural information resolved by the ILP is limited for larger RNAs, we decided to obtain better insight into the ModT structure both in vitro and in vivo using dimethyl sulfate mutational profiling (DMS-MaP) [30,31]. DMS-MaP is a well-established method to infer secondary structure by exploiting DMS reactivity at single stranded, or exposed, A and C nucleotides. We generated DMS-reactivity profiles of ModT(S) and ModT(L) under various conditions: (1) in vitro transcribed ModT isoforms; (2) in vivo, from cultures grown to exponential, transition, and stationary phase; and (3) from deproteinated cell lysates obtained at the same growth phases (Fig 2D). Of note, we specifically determined the reactivity values of the long isoform (L) using a primer binding to the extended 3′-end for reverse transcription in vivo and in deproteinated cell lysates. As this is not possible for the short isoform, reactivities of these samples (S) represent a combined contribution of ModT(S) and ModT(L), which might mask minor structural differences.
Importantly, DMS reactivities for both ModT isoforms showed very high correlations (≥0.98) in each analyzed condition (S2E Fig), with no prominent differences between ModT(L) and ModT(S) within the common region (Fig 2D). This is particularly apparent when subtracting their normalized reactivities (Fig 2E). Hence, the DMS probing confirmed the ILP data, indicating that ModT(L) and ModT(S) adopt the same core structures in vitro and in vivo. Generally, we noticed a strong positive correlation across all analyzed samples (≥0.86), which indicates overall similar RNA structures for ModT in different growth conditions (S2E Fig). Nevertheless, hierarchical clustering using the single linkage algorithm showed that the samples from the late time points display the lowest reactivity across several regions, while those from the exponential phase (EP) have an overall increased reactivity (Fig 2D). Interestingly, higher overall reactivities were also observed for the Proteinase K digested lysates and for the in vitro transcribed RNA samples (Fig 2D). Arguably, the key difference between the native in vivo conditions, and the in vitro/deproteinated conditions, respectively, is the absence of proteins in the latter two, which indicates the interaction of ModT with proteins in vivo (Figs 2D and S2E).
Specifically comparing reactivity changes of single residues between conditions (e.g., in vivo deproteinated versus in vivo native, (Figs 2E and S2F) identified strong changes in base-paired regions P7 and P8, and in the regions with the lowest structural confidence across the 3 ModT structure models (P3-P5) (Fig 2A–2C). Therefore, we wanted to explore whether our experimental DMS-MaP data of ModT could identify a model with best fit to the ModT structural data. For that, we calculated the ROC-AUC score (receiver operator characteristic–area under the curve), a summary statistics, which serves as a measure of the agreement between the predicted base pairing in a model and the experimentally determined DMS reactivity [31]. Indeed, this analysis clearly favored the updated consensus secondary structure model (Fig 2A), as it displayed the highest ROC-AUC score across all tested conditions (S2G Fig).
In summary, our data suggest overall identical structures for both isoforms in vivo and in vitro. Furthermore, the identification of a best-fitting structure model provides a crucial step in delineating the structural constraints for ModT function as it will guide the design of mutations in critical base-paired regions in future experiments.
ModT acts in trans and regulates transition into the stationary phase of growth
As a first step towards identifying cellular functions of ModT, we deleted the first 215 nt of modT (ΔmodT) corresponding to the conserved ModT(S) isoform (Fig 3A). This abolished production of both isoforms without causing any polar effects on neighboring genes (CDIF630_00250 and CDIF630_00251) (S3A and S3B Fig). Due to the nutrient-dependent expression profile of ModT, we initially compared growth of wild-type (WT) and ΔmodT in TY medium supplemented with carbon and energy sources that can be utilized by C. difficile during gut colonization (Fig 3B) [32–34]. The deletion of modT was associated with an increased maximum optical density (OD) (S3C Fig), which appeared to be the result of extended exponential growth (Fig 3B). This overgrowth phenotype was particularly pronounced in media containing hexose monosaccharides (glucose, fructose, and mannose) or ethanolamine. Curiously, the overgrowth did not correlate with the carbon source-dependent ModT expression in late stationary phase, which does not occur during growth on ethanolamine (Fig 1C). Nevertheless, it indicates that the presence of ModT in exponential phase of growth is needed for a timely transition into the stationary phase. Importantly, in trans chromosomal complementation of ΔmodT with either the short or the long isoform resulted in an expression level and pattern similar to wild-type ModT (S3B Fig) and restored growth to wild-type level for both complementation strains (Fig 3B). This demonstrates that the short isoform is sufficient for ModT function. Furthermore, and in agreement with a recent study in C. acetobutylicum [22], this experiment rules out a cis-acting riboswitch function of ModT, which was originally proposed based on its genetic location and complex secondary structure [15].

Fig 3. Growth analysis and transcriptomic profiling of modT mutants reveal a trans-acting function important for the transition into the stationary phase.
(A) Schematic of chromosomal modT deletion, and in trans complementations at the pyrE locus of the short and the long isoforms, respectively. (B) Growth curves of C. difficile 630 WT, ΔmodT and complemented strains in TY medium supplemented with indicated carbon sources. The underlying data can be found in S5 Dataset. (C, D) Volcano plots showing transcriptomic comparison between C. difficile 630 WT and ΔmodT grown in TYG to EP (3.5 h) and TP (9 h). FDR cutoff was set to 35] are indicated in red, genes induced during sporulation in a Spo0A-dependent manner [39] are labeled blue. Up-regulated tRNA genes are colored in purple. The underlying data can be found in S2 and S5 Datasets. EP, exponential phase; FDR, false discovery rate; TP, transition phase; WT, wild type.
To identify cellular pathways that are regulated by ModT, we performed comparative transcriptome analysis. Wild-type and ΔmodT strains were grown in TY medium supplemented with glucose (TYG). We collected samples in the EP (3, 5 h) and the TP (9 h), when the knockout strain begins to outgrow the wild type (S3D Fig). This analysis identified 145 differentially regulated genes in the exponential phase and 344 differentially regulated genes in the TP (each with a false discovery rate (FDR) ≤ 0.1 and a log2 FC ≥ 1 or ≤ -1) (Figs 3C, 3D, and S3E and S2 Dataset). The list of regulated genes was dominated at both timepoints by metabolic genes and ABC-type sugar and amino acid transporters, which might be linked to the observed carbon-source dependence of ModT expression and the growth phenotype of the mutant.
Given the outgrowth phenotype of ΔmodT, we focused on differentially expressed genes in the TP. We noticed that deletion of modT was associated with broad down-regulation of the σB-dependent regulon. Sigma B regulates the general stress response in C. difficile, mediating tolerance towards many stress conditions, but also adaptive responses during intestinal colonization [35,36]. In our dataset, approximately 25% (62/248) of the genes that are under positive control of σB [35], were significantly down-regulated in ΔmodT during the TP (S3F Fig and S3 Dataset). In accordance with a low σB activity during early exponential phase [36], only 7% of the regulon (16 genes) was down-regulated at this time point.
We further observed down-regulation of genes under the control of σH, a central regulator of the TP [37]. Its regulon is less defined than the σB regulon, but of the 40 genes known to harbor a σH promoter [37], 18 (45%) were down-regulated in ΔmodT during the TP (S3 Dataset). Furthermore, we found globally increased tRNA abundance in the mutant, with 75 out of 90 tRNA loci being up-regulated. Given the technical challenges associated with tRNA isolation and sequencing [38], we sought to validate this observation by northern blotting. To that end, we generated a high-resolution time curve covering all growth stages of C. difficile and probed for 2 different tRNAs (S3G Fig). This validated the increased abundance of these tRNAs in the knockout during the TP, in comparison to the wild type. Interestingly, the difference increases at late stationary phase time points due to a reduction of the tRNA fraction in the wild type, while levels are stably maintained in the knockout. Intriguingly, probing for 5S indicated that this trend also occurs with ribosomal RNA.
In addition to a broad down-regulation of the σB and σH regulons, we identified pathways that directly regulate toxin production and sporulation. Specifically, we observed up-regulation of the tcdA gene, encoding for the large clostridial toxin A (log2FC = 1.1 and 0.9 in exponential and transition phase, respectively), and of the tcdR gene (log2FC = 2.3 in TP), encoding for the alternative sigma factor TcdR that positively controls synthesis of the toxin genes tcdA and tcdB. This indicated that ModT activity may limit toxin production. To test whether the observed transcriptome changes translate into higher toxin levels, we compared these between wild type and mutant. This revealed that the modT mutant produced more toxin than the wild type (Fig 4A, left panel), which ultimately resulted in higher levels of toxin released into the culture supernatant (Fig 4A, right panel). Importantly, as seen for the growth phenotype, complementation of either the short or the long isoform of ModT restored toxin production to wild-type levels.

Fig 4. ModT regulates stationary phase associated spore formation and toxin production.
(A) ELISA-based toxin quantification (TcdA and TcdB combined) in cell lysates (“intracellular toxin”) and cell free supernatant (“secreted toxin”) of C. difficile 630 WT, ΔmodT and complemented strains grown in TYG medium. Relative toxin amount is displayed as absorbance at 450 nm (A450) normalized by the absorbance at 620 nm (A620). n = 3 biological replicates. The underlying data can be found in S5 Dataset. (B) Representative phase-contrast micrographs (n = 3 biological replicates) of C. difficile 630 WT, and modT mutants in 70:30 medium. Time points are indicated relative to the onset of the TP. Scale bar = 5 μm. (C) Sporulation frequency calculated from the phase-contrast microscopy displayed in (B). n = 3 biological replicates. Time points are indicated relative to the onset of the TP. The underlying data can be found in S5 Dataset. Significance for data shown in (A) and (C) was determined by two-way ANOVA with Tukey’s multiple comparisons test. Only significant differences with P P P P
Furthermore, 23 sporulation genes [39] were down-regulated in TP (Fig 3D and S3 Dataset), including those encoding for the sporulation sigma factors σE (log2FC = −1.1), σF (log2FC = −0.92), and σG (log2FC = −1.05), all of which coordinate spore developmental steps downstream of the master regulator of sporulation Spo0A. Therefore, we quantified sporulation frequencies in wild-type, ΔmodT and complemented strains over an extended period of 5 days. Spore enumeration by phase contrast microscopy revealed a significant decrease in sporulation frequency in the knockout (Fig 4B and 4C), which was independently validated by CFU-based calculation of vegetative cells and ethanol-resistant spores (S4A and S4B Fig). This sporulation defect remained throughout the experiment, indicating that sporulation is not just delayed in ΔmodT, but permanently repressed. Surprisingly, while chromosomal complementation of ΔmodT with the long isoform (ΔmodT C-long) reconstituted sporulation to wild-type levels, complementation with the short isoform (ΔmodT C-short) resulted in a pronounced hyper-sporulation phenotype (Figs 4B, 4C, S4A, and S4B). Northern blot validation of ModT revealed both a mild increase in ModT(S) transcript levels and a prolonged expression of ModT(S) at later time points in the short complementation strain, relative to the WT or ΔmodT C-long (S4C Fig). This either suggests that small alterations in ModT abundance and/or presence of ModT in stationary phase can have a strong impact on sporulation, or that the 2 isoforms differ in their capacity to regulate sporulation.
Taken together, the extended exponential growth and the global down-regulation of the σB and σH regulons in ΔmodT indicates that ModT regulates the transition from exponential to stationary phase of growth in C. difficile. Concomitantly, we observe changes in spore formation and toxin production, 2 virulence-determining processes that are tightly regulated in response to the physiological state of the cell.
ModT regulates sporulation by modulating cellular cyclic di-GMP levels
We sought to better understand the mechanistic basis for the observed changes in toxicity and sporulation in the modT mutant. Our transcriptome analysis highlighted several differentially expressed genes shown to affect sporulation (highlighted in Fig 5A). Among them, the dccA gene caught our attention, because it was the top up-regulated transcript at both time points in ΔmodT (Fig 3C and 3D and S2 Dataset). DccA is a diguanylate kinase containing a GGDEF domain that catalyzes the synthesis of the second messenger c-di-GMP [40]. Overexpression of DccA was shown to suppress early sporulation events, downstream of Spo0A, by increasing cellular c-di-GMP levels [41] through a yet unknown mechanism. Increased dccA transcription, and consequently, increased c-di-GMP levels, have been associated with a sinRR’ deletion mutant which is asporogenous and displays global gene expression changes in pathways regulating sporulation [42]. Aligning with that, the bicistronic operon encoding SinR and its protein antagonist SinR’ [42], was down-regulated in ΔmodT at both time points (Fig 5A). Hence, our transcriptome analysis of the modT mutant (S2 Dataset) suggested that the pronounced effects on sporulation could be linked to changes in cellular levels of c-di-GMP.

Fig 5. ModT modulates sporulation and toxin production via changes in cellular c-di-GMP levels.
(A) Schematic overview of differentially expressed genes in ΔmodT that were shown to be involved in the regulation of spore formation in C. difficile [40–42,56,65]. (B) Changes in c-di-GMP levels in ΔmodT and in complemented strains grown in TYG medium. Measurements were done using a fluorescent c-di-GMP type II riboswitch reporter. Values in the ΔmodT and in complemented strains were calculated as fold-change relative to the C. difficile 630 WT signal. Data represent the mean of 3 independent experiments performed with n = 3 biological replicates per experiment (total n = 9). The underlying data can be found in S5 Dataset. (C) c-di-GMP levels in WT, ΔmodT and ΔmodTΔdccA-01583. Measurements were done as in (B) Values in the ΔmodT and ΔmodTΔdccA-01583 strains were calculated as fold-change relative to the C. difficile 630 WT signal. The underlying data can be found in S5 Dataset. (D) Assessment of sporulation frequency in 70:30 medium by phase-contrast microscopy. The underlying data can be found in S5 Dataset. (E) Relative quantification of toxin (TcdA and TcdB combined) as determined by ELISA in cell free supernatants of C. difficile 630 WT, ΔmodT and ΔmodTΔdccA-01583 grown in TYG medium. Each experiment was performed in 3 (n = 3) biological replicates. The underlying data can be found in S5 Dataset. For the experiments shown in (B) and (C), the fluorescence values were normalized to the control (pRSA>G) as described in S5B Fig. For data shown in (B) and (C), significance was determined by two-way ANOVA with Tukey’s multiple comparisons test; for data shown in (D) and (E), one way ANOVA with Tukey’s multiple comparisons test was applied. Only significant differences with P P P P
To investigate this link, we assessed whether deletion of ModT was associated with increased cellular c-di-GMP levels. For this, we adopted a reporter system in which a c-di-GMP sensitive riboswitch represses transcription of a downstream encoded mCherry protein at low c-di-GMP levels [43–45] (S5A Fig). Hence, increasing mCherry fluorescence serves as a proxy for increased c-di-GMP levels. To control for background fluorescence, we employed a reporter with an A70G mutation (pRSA70G) that renders the riboswitch unresponsive to c-di-GMP. Wild-type, ΔmodT and the complemented strains, each harboring the different reporter constructs and an empty vector control, were grown in TYG medium to mid-exponential (6 h) and stationary phase (24 h), and mean fluorescence intensities were assessed by flow cytometry (S5B Fig). Comparing the mean fluorescence in each strain background carrying the functional reporter (pRS), the mutated reporter (pRSA70G), and the empty vector control (pEV), revealed some residual fluorescence activity for the mutated reporter (S5B Fig). Therefore, all fluorescence data were normalized to the background fluorescence of the same strain carrying the mutated reporter construct (Fig 5B). There were no overall significant differences between the WT and the ΔmodT strain in the early stages of growth. However, at 24 h, we consistently detected increased fluorescence in the modT mutant. Importantly, this increase in reporter activity could be reverted to WT levels in both complemented ModT strains (Fig 5B). This confirmed our initial hypothesis that deletion of modT leads to increased cellular c-di-GMP levels, presumably through increased levels of DccA.
The dccA gene is co-transcribed with CDIF630_01583, which is a putative diguanylate kinase signaling protein. However, it also contains several potential sensory domains of the Per-Arnt-Sim (PAS) type, in addition to an EAL-domain which may degrade c-di-GMP [46]. The regulatory impact of CDIF630_01583 has not been studied so far. Since the entire operon was up-regulated in our dataset, we wanted to test if and how CDIF630_01583-dccA up-regulation was contributing to the changes in sporulation in the modT mutant. For that, we deleted the dccA-01583 operon in ΔmodT and compared the fluorescence of the resulting double knockout to ΔmodT and the wild type. This revealed that c-di-GMP levels did not increase in a ΔmodT ΔdccA-01583 double knockout, and even dropped below WT levels (Fig 5C). Consequently, deletion of the dccA-01583 operon in the ΔmodT strain background did not just restore sporulation frequency to WT levels, but actually increased the frequency (Fig 5D). Taken together, these data show that modT deletion leads to sporulation inhibition via increased DccA-mediated c-di-GMP synthesis. In contrast to sporulation, we observed only partial restoration of toxin production in the ΔmodT ΔdccA-01583 double knockout (Fig 5E), suggesting additional, c-di-GMP-independent, regulatory mechanisms contributing to the observed increases in toxin production in ΔmodT.
In summary, the effects of ModT on spore formation are mediated through the ubiquitous second messenger c-di-GMP.
ModT orthologs from distant phylogenetic families are functionally conserved
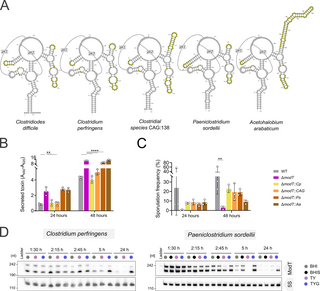
Given the high sequence conservation of ModT family members, we wanted to explore whether ModT orthologs from diverse species are capable of complementing ModT function in C. difficile. To this end, we exchanged the sequence of the consensus ModT motif (first 215 nt) in C. difficile (S6A and S6B Fig) with the consensus motif of 3 ModT orthologs from the phylum Bacillota, including Clostridium perfringens and unclassified Clostridium sp. CAG:138 (genus Clostridium, family Clostridiaceae), as well as Paeniclostridium sordellii (genus Paeniclostridium, family Clostridiaceae). In addition, we chose 1 ortholog from the phylum Actinomycetota, Acetohalobium arabaticum (genus Acetohalobium, family Halobacteroidaceae). All 4 orthologs possess core structural features of the ModT family, but vary in size and sequence, particularly in the regions of the variable P5, P7, and P8 stems (Figs 6A and S6A). For example, while the P7 and P8 stem regions comprise only ~51 nt in C. difficile, they are 73 nt and 118 nt long in CAG:138 and A. arabaticum, respectively. Furthermore, the primary sequences of the P5 loop vary in all strains and contain covarying mutations in various places. Northern blot validations of ModT transcripts in complemented strains showed that the orthologs from C. perfringens and CAG:138 are expressed at levels similar to WT ModT, although the ratios between short and long isoform vary (S6C Fig). In contrast, strains expressing orthologs from P. sordellii and A. arabaticum showed low ModT levels (S6C Fig). Despite expression level differences, we assessed toxin production and sporulation frequency in all complemented strains to determine whether these orthologs can replace ModT function in C. difficile. In comparison to the ΔmodT mutant, the orthologs of C. perfringens and CAG:138 were able to fully restore both toxin production and sporulation frequency to WT levels (Figs 6B, 6C, and S6D–S6F). P. sordellii ModT was able to restore sporulation frequency, whereas toxin levels remained comparable to ΔmodT. Similarly, complementation with ModT from A. arabaticum was able to partially rescue sporulation but did not restore toxin production. The fact that P. sordellii and A. arabaticum orthologs can only partially rescue the ΔmodT associated phenotypes might simply be due to their weaker expression in C. difficile, but it could also be an indication for a functional divergence.

Fig 6. Orthologs from distant phylogenetic families can replace ModT function in C. difficile.
(A) Consensus secondary structure models of ModT orthologs used for complementation. Regions varying in primary sequence and secondary structure across all orthologs are highlighted in yellow. (B) Relative quantification of toxin (TcdA and TcdB combined) determined by ELISA in cell free supernatant of WT, ΔmodT and C. difficile with ModT ortholog replacements grown in TYG medium. Each experiment was performed in 3 (n = 3) biological replicates. The underlying data can be found in S5 Dataset. (C) Assessment of sporulation frequency in 70:30 medium based on phase-contrast microscopy images (n = 3 biological replicates) shown in S6D Fig. The underlying data can be found in S5 Dataset. (D) Northern blot-based expression analysis of ModT in C. perfringens and P. sordellii across different growth stages in various rich media, including BHI, BHI + yeast extract (BHIS), Tryptone Yeast (TY), and TY supplemented with 0.5% glucose (TYG). 5S rRNA served as loading control. Depicted is a representative of 3 biological replicates. Significance for data shown in (B) and (C) was determined using two-way ANOVA with Dunnett’s multiple comparisons test. Only significant differences with P P P P
To develop a better understanding of whether ModT orthologs carry out conserved functions, we sought to explore their expression and isoform profiles within their native hosts. For that purpose, we chose the 2 human pathogens Clostridium perfringens and Paeniclostridium sordellii that belong to the family Clostridiaceae. Although they are phylogenetically distant from C. difficile (family Peptostreptococcaceae), they are both spore formers and encode homologs of the large clostridial toxins TcdA and TcdB [47,48]. Both bacteria were grown in media with varying levels of glucose to collect time-resolved expression profiles across the different growth stages. This expression analysis revealed several interesting parallels to C. difficile ModT (Fig 6D, compare to Fig 1C). Firstly, both orthologs occur in 2 isoforms, one matching the conserved ModT motif (~210 nt) and a larger isoform of ~240 nt. Secondly, modT is also strongly expressed during exponential growth, but depleted in the stationary phase. Third, medium supplementation with glucose extends the expression of the RNAs into the stationary phase. Although a recent expression analysis of ModT in C. acetobutylicum did not analyze the effect of different carbon sources on ModT expression, it also showed that ModT transcript levels decrease after entry into stationary phase [22]. In addition, the promoter regions of modT in all 4 clostridial species contain a highly conserved sigA consensus motif (S6G Fig), strongly suggesting that SigA-mediated modT expression in the exponential phase is widespread and may be a key aspect of its regulation. Despite these similarities, there is one major difference in that ModT(S) isoforms appear more stable than the ModT(L) isoforms in C. perfringens and P. sordellii, while the long isoform is lacking entirely in C. acetobutylicum.
In summary, the conserved expression profiles in various bacteria, along with the capacity of orthologs to complement ModT activity in C. difficile suggests that ModT members from diverse phylogenetic backgrounds are functionally conserved.
Discussion
Broadly conserved, structured noncoding RNAs are central regulators of fundamental cellular processes in bacteria. Our characterization of the ModT RNA in C. difficile echoes this notion as its deletion results in pronounced phenotypes under standard growth conditions. These comprise delayed transition from the exponential to the stationary phase of growth, reduced spore formation, and increased toxin production. Our results align with a recent study, which showed that deletion of modT results in sporulation and aggregation deficiency in C. acetobutylicum [22]. These findings strongly suggest a conserved role for ModT in regulating lifestyle transitions in different bacterial species.
Here, we show that ModT regulates this lifestyle transition, at least in part, through modulating cellular levels of the second messenger molecule c-di-GMP. We show that deletion of modT is associated with increased cellular levels of c-di-GMP, and we were able to causally link this increase in c-di-GMP to the observed sporulation defect in the modT mutant. Our results complement previous findings that the overexpression of dccA, and associated increases in cellular c-di-GMP, lead to significant decreases in sporulation frequencies [41]. These findings are intriguing because c-di-GMP is a nearly ubiquitous bacterial second messenger that regulates fundamental processes such as bacterial growth and behavior (motility, aggregation, and biofilm formation), but also developmental processes, such as spore formation, in diverse bacteria [49].
In fact, regulation of c-di-GMP levels by ModT might explain the sporulation and aggregation defects observed in a modT mutant in C. acetobutylicum [22], as both are known c-di-GMP-dependent processes in bacteria [49]. In the future, it will be interesting to see if ModT orthologs generally impact biological processes that are regulated by c-di-GMP.
Toxin production in C. difficile is also a c-di-GMP-dependent process, but the current model for c-di-GMP mediated regulation of toxin production [50,51] does not align with the increased toxin production in the modT mutant. That is, c-di-GMP was shown to negatively regulate toxin production by mediating the riboswitch-dependent premature transcription termination of the flagella operon at high c-di-GMP levels. As a consequence, sigD, which drives TcdR production and which is encoded in the flagella operon, is repressed. In our transcriptome analysis, we do not observe such down-regulation of the flagellar operon, which suggests that the toxin phenotype is driven by c-di-GMP-independent changes in the modT mutant. In line with this, we observed only partial restoration of toxin production in the ΔmodT ΔdccA-01583 double knockout. However, we would like to point out that sporulation assays and toxin quantification were performed in different media. Thus, to dissect the relative contribution of ModT-mediated changes in c-di-GMP levels to toxin production and spore formation, and to understand how both processes effect each other in the modT mutant, further experiments are required, e.g., under conditions when both sporulation and toxin production occurs.
Curiously, the identified stationary phase phenotypes of modT mutant strains (sporulation, aggregation) seem to be inversely correlated with the largely conserved expression of ModT during the exponential phase of growth in several clostridial species. In fact, C. difficile ModT is among the most abundant and most stable RNAs in the cell during vegetative growth, both in C. difficile and C. acetobutylicum. However, we show that its levels drop almost completely upon transition of bacteria into the stationary phase in C. difficile, C. perfringens, and P. sordellii. This suggests that the presence of ModT might be needed to initiate the timely transition from exponential to stationary phase of growth. In this context, the ModT mediated phenotypes could be interpreted as a failure to sense and/or transmit a stress signal, resulting in the observed prolonged vegetative growth of the mutant. The sporulation defect might subsequently result from a lack of energy to produce spores. Likewise, the up-regulation of toxin production could indicate an inadequate coordination between central metabolic pathways and the regulation of toxin genes in the modT mutant.
An interesting feature of ModT that stood out to us is the existence of a longer isoform that differs from the short isoform through a 3′ extension. Due to a lack of primary sequence conservation in the extended 3′-end, it was missed in the original genome-based annotation by Weinberg and colleagues [15]. Based on our data, we propose that ModT(L) is the result of read-through transcription into an intrinsic terminator downstream of ModT(S) in C. difficile. This longer isoform does not appear to be specific to C. difficile, because we detected it in C. perfringens and P. sordellii as well. That said, a recent expression analysis of the ModT ortholog in C. acetobutylicum [22] did not reveal the existence of a long isoform, which does raise the question how common it might be. To gather some clues, we revisited the original conservation analysis [15] which contained in silico terminator predictions for orthologous ModT sequences, including C. difficile. These analyses predict rho-independent transcription terminators in close proximity to the 3′-end of ModT(S) for all 3 clostridial species expressing a long ModT isoform but not for ModT from C. acetobutylicum ([15], supplemental files). Rho-independent transcriptional terminators are complex structures that comprise both a stem-loop and a poly-uridine stretch which suggests that they do not evolve easily. Therefore, it seems more plausible that intrinsic terminators were originally present in all ModT orthologs, but have been lost over time in individual bacteria.
Are the 2 isoforms just the result of transcriptional read-through events or do they confer distinct functions in those organisms where they are produced? Although this needs to be carefully investigated in the future, our analyses in C. difficile provide initial evidence for potential functional divergence between both isoforms. That is, although our structure probing data suggest that both isoforms adopt a highly similar structure, the in vivo half-lives differ markedly between ModT(L) and ModT(S). The increased stability of ModT(L) suggests that the stable secondary structure of the terminator mediates protection against 3′–5′ exonuclease activity [52]. However, it could also indicate engagements of both isoforms in different ribonucleoprotein complexes. Furthermore, complementation with the short, but not with the long isoform, leads to a striking hyper-sporulation phenotype, even though both isoforms restore c-di-GMP to a level comparable to the WT. This suggests that ModT(S) might carry out additional roles that impact sporulation frequency in a c-di-GMP independent manner, possibly through interactions with other protein regulators. This hypothesis could be tested in the future, by performing isoform-specific pulldowns of ModT to identify interacting protein and RNA partners.
But what is the underlying molecular function of ModT? The broad sequence conservation across the entire ModT RNA implies a function that is not linked to the simple base-pairing mechanism of small regulatory RNAs. Their primary sequence conservation is typically very narrow, and mostly order or genus specific [8]. Sequence conservation is usually restricted to the seed region of complementarity to their regulated target mRNAs. Furthermore, our previous analyses of the two known sRNA-binding proteins in C. difficile, Hfq and KhpB, did not reveal binding of ModT to either of these 2 global RBPs [21,26,53]. Therefore, based on our current knowledge, it seems unlikely that ModT acts as a canonical sRNA.
Broadly conserved bacterial ncRNAs with intricate secondary and tertiary structure, such as ModT, often act as riboswitches or ribozymes. Our study confirms recent findings in C. acetobutylicum that phenotypes of modT knockout strains can be complemented in trans through expression of modT from a plasmid or an independent genomic locus, thereby ruling out a cis-acting riboswitch function for ModT [22]. These data, however, do not exclude the possibility that ModT has evolved from an ancient riboswitch to carry out trans-regulatory functions. Curiously, our in vivo DMS-MaP data show changes in reactivities in ModT between the exponential and the transition/stationary growth phases which opens the possibility that ModT still acts like a “riboswitch” in that it binds a ligand which induces conformational changes in ModT. Binding assays with potential ligands, such as c-di-GMP, combined with DMS-MaP analyses should provide more insights into this hypothesis in the future.
Other conserved RNA classes with intricate tertiary structures are catalytic RNAs, called ribozymes [5]. Almost all described ribozymes are self-cleaving RNAs, the only exception is RNase P which acts on other RNA substrates, and is involved in pre-tRNA processing [9]. However, in light of the increased levels of tRNAs and 5S RNA in the modT knockout strain, it is an intriguing, yet speculative, thought that ModT might be involved in the processing of other RNAs, including tRNAs and rRNAs. Therefore, future experimental efforts should focus on both potential self-cleaving and trans-cleaving activities of ModT. Some ribozymes, such as the glmS ribozyme, catalyze RNA cleavage in response to binding a specific metabolite [54]. Therefore, these cleaving assays should be performed in the presence of potential ligands such as c-di-GMP.
Interestingly, in about approximately 20% of all ModT species (including C. difficile), the P8 stem displays an E-loop structure [15,22], that also occurs in ribosomal RNAs as recognition sites for RNA–protein interactions. This may indicate that at least some ModT orthologs engage in protein interactions via this structural element [55]. Support for this hypothesis comes from our previous Grad-seq (gradient profiling by sequencing) analysis in C. difficile 630, to predict RNA and protein complexes based on glycerol gradient sedimentation profiles [26]. This revealed sedimentation of both ModT isoforms in molecular weight fractions that indicate their association with a protein (or protein complex). Furthermore, our DMS-structure probing analysis indicated increased reactivities in several regions of the RNA in deproteinated versus native cell lysates. Our current selection of ModT orthologs for functional complementation of the modT knockout in C. difficile did not contain an ortholog lacking the E-loop motif. Hence, in the future it will be interesting to determine whether orthologs lacking this motif can equally complement all phenotypes in C. difficile, and whether this motif contributes to RNA-protein interactions.
In summary, our findings establish ModT as a conserved regulator of the bacterial life cycle and provide experimental avenues towards delineating the molecular mechanism of its intricate RNA structure.
Materials and methods
Bacterial strains and growth conditions
A list of bacterial strains used in this study is provided in S4 Dataset. C. difficile strain 630 (DSM 27543) cultures were routinely grown at 37°C inside a Coy chamber (85% N2, 10% H2, and 5% CO2) on brain heart infusion (BHI) agar plates (1.5% agar) or in autoclaved BHI broth (37 g/l). Petri dishes were anaerobically incubated for at least 4 h prior to inoculation, while liquids were kept in the Coy chamber overnight to allow diffusion of entrapped oxygen. For selection of plasmid-harboring C. difficile 630, antibiotics were added at the following concentrations: thiamphenicol 15 μg/ml, cefoxitin 8 μg/ml, cycloserine 250 μg/ml. Cryostocks were stored at −80°C in BHI broth containing 15% glycerol.
To cultivate C. difficile with different carbon and energy sources autoclaved TY medium (30 g/l tryptone (Roth), 20 g/l yeast extract (Roth)) was supplemented with the respective source from 20% (w/V) sterile filtered stock solutions in ddH2O to a final concentration of 0.5%. For growth curves, TY day cultures were inoculated to an OD600 of 0.05 from TY overnight cultures, grown to exponential phase (OD600 ~0.5) and subsequently diluted to an OD600 of 0.01 in TY containing the respective carbon source. For growth curves in BHI and BHIS medium (BHI with 5 g/l of yeast extract (Roth)), cultures were inoculated from exponential phase BHI cultures. Optical density at 600 nm was recorded in a transparent 96-well plate in a Biotek H1 microplate reader.
C. perfringens ATCC 133124 and P. sordellii ATCC 9714 were cultured under the conditions indicated for C. difficile 630, with the following changes: For growth of P. sordellii in BHIS medium, 10% w/V sterile filtered cysteine solution was added to a final 0.1% (w/V) after autoclaving. For C. perfringens, the overnight culture was replaced with a day culture. Escherichia coli (E. coli) strains were aerobically cultivated in autoclaved Luria–Bertani (LB) broth (10 g/l tryptone, 5 g/l yeast extract, 10 g/l NaCl) at 37°C with shaking at 220 revolutions per minute (RPM), or on LB-agar (1,5% agar). Plasmids were maintained by adding 20 μg/ml chloramphenicol as a selection antibiotic. Cryostocks were stored at −80°C in LB broth with 25% glycerol.
Toxin ELISA
For C. difficile toxin quantification, 2.5 OD600 units of bacterial culture were centrifuged at 10,000 × g, 4°C for 10 min. The supernatant was transferred to a new tube and centrifuged again under the same conditions to remove any residual cells. Approximately 250 μl cell free supernatant was stored at −80°C until quantification of the “secreted toxin.”
The cell pellet (used for quantification of “intracellular toxin”) was washed with 1 ml PBS (phosphate-buffered saline), then centrifuged again to remove the remaining liquid. Samples were kept at −80°C until further processing.
Cell pellets were suspended in 500 μl of ddH2O and transferred to a new tube containing 500 μl 0.1 mm glass beads. For lysis, samples were ground in pre-cooled adapters (−20°C) in a Retsch Mixer Mill at 30 Hertz for 10 min, and subsequently centrifuged at 10,000 × g, 4°C for 15 min to remove debris and beads; 200 μl of the clear supernatant was transferred to a fresh tube and the protein concentration was estimated with a Bradford assay using RotiNano Quant solution (Roth) according to the manufacturer’s instructions. For relative toxin quantification, 4 μg of protein from the “intracellular toxin” sample were used as input, while “secreted toxin” was assessed from 50 μl of the cleared supernatant. The relative amount was determined by toxin ELISA (TGC-E001-1, TGC Biomics) performed and evaluated according to the manufacturer’s instructions. ELISA signal was quantified with a Tecan infinite 200 Pro plate reader assessing absorption at 450 nm and 620 nm. As a final readout, the signal at 620 nm was subtracted from the 450 nm signal.
Sporulation assays
Sporulation assays were performed in 70:30 sporulation broth medium according to Edwards and colleagues with minor alterations [56]. Overnight TY cultures were used to inoculate day cultures to an OD600 of 0.05 in BHIS containing 0.5% fructose and 0.1% taurocholate. After growth to mid-exponential phase, 70:30 medium was inoculated to an OD600 of 0.01.
At the end of transition phase (approximately 8 to 10 h after inoculation), the total CFUs from untreated cultures were assessed by serial dilution and spotting in BHIS, and in addition, an aliquot treated with ethanol for 30 min was serially diluted on BHIS agar with 0.1% taurocholate to check for spore carryover. This time point is referred to as 0 hours. At the subsequent time points, vegetative CFUs were assessed by serial dilution and plating on BHIS agar and spores were quantified from culture treated with ethanol (EtOH) (final concentration 48% v/V) for 30 min, followed by serial dilution in PBS and plating on BHIS containing 0.1% taurocholate.
For a second independent analysis at all time points, 500 μl culture aliquots were concentrated by brief centrifugation and approximately 3 μl of the concentrated cells were spotted onto an agarose pad on a glass slide for microscopy. Spores were visualized by phase contrast microscopy using a HC PLAN FLUOTAR 100×/1.32 PH3 oil immersion objective on a LEICA DM2500 microscope. Two fields of view were acquired per biological replicate from which at least 300 cells per field were counted to determine the sporulation frequency. In both methods, sporulation frequency was calculated as [spore count] / [spore count + vegetative cell count] = sporulation frequency.
Transformations
Plasmids were transformed to E. coli Top10 by applying a heat shock for 1 min at 42°C. After chilling on ice for 2 min, bacteria were incubated at 37°C, 350 RPM for 1 to 4 h in LB medium without antibiotics and were subsequently plated on selective LB-agar.
For transformations into E. coli CA434, 80 μl of a bacterial suspension was thawed on ice and mixed with 1 to 2 μl plasmid in a pre-chilled electroporation cuvette. Plasmid was delivered with an electric pulse (1.8 kV, 200 Ω, 4–5 s). Bacteria were recovered at 37°C in LB broth at 350 RPM for 1 h and subsequently streaked on selective LB agar.
Plasmid conjugation to C. difficile 630
For conjugations, 1 ml of an E. coli CA434 overnight culture harboring the respective plasmid was centrifuged at 4,000 × g, RT for 2 min. The pellet was transferred into the Coy chamber, and resuspended in 200 μl overnight culture of the respective C. difficile strain. After spotting onto BHI agar and 8 h of incubation at 37°C, cells were scraped from the plate with approximately 700 μl BHI broth and restreaked on BHI agar containing antibiotics to select for bacteria harboring the plasmid. Colonies appeared after 24 to 72 h and were restreaked to purity on selective BHI agar plates.
Plasmid assemblies
All used and generated plasmids are listed in S4 Dataset.
Deletion of modT in C. difficile 630 (pFF-173).
For allele exchange cassettes, approximately 1.2 kB of the genome regions flanking the 215 nt long knockout site were PCR-amplified from C. difficile 630 genomic DNA with FFO-923/926 and FFO-924/927. Fragments were merged by SOEing PCR, gel-purified and subsequently digested using SacI (Thermo Scientific) and BamHI (Thermo Scientific). The fragment was ligated into equally digested, gel-purified pJAK112 [21] using T4 DNA ligase (Thermo Scientific), yielding pFF-173.
In trans-complementations of C. difficile 630 modT into the neutral pyrE locus (pFF-221/222).
For chromosomal complementation into the pyrE-locus, modT including the native promoter sequence was amplified from C. difficile 630 gDNA using FFO-1088/1034 (short isoform) or FFO-1089/1034 (long isoform). The vector backbone was amplified from pJAK080 [21] using FFO-1086/1087, which included the sequence of the slpA terminator to prevent read-through transcription of modT upon chromosomal integration. The vector and inserts were digested using KpnI (Thermo Scientific), gel-purified and subsequently ligated with T4 DNA ligase resulting in the plasmids pFF-221 (short isoform) and pFF-222 (long isoform).
Replacement of C. difficile 630 modT with orthologues (pFF-314/320/321/377).
To replace the short C. difficile 630 modT sequence in its native locus, orthologue sequences were PCR-amplified from the respective species’ genomic DNA, using FFO-1294/FFO-1297 for C. perfringens ATCC13124, FFO-1443/1444 for P. sordellii ATCC 9714, FFO-1305/1306 for Acetohalobium arabaticum DSM 5501 and FFO-1503/1521 for the Clostridium species CAG138. Up- and downstream allele exchange cassettes were amplified from C. difficile 630 genomic DNA using FFO-923/1295 and FFO-924/1296 for C. perfringens, FFO-923/1445 and 924/1442 for P. sordellii, FFO-923/1303 and FFO-924/1304 for A. arabaticum, and FFO-923/1502 and FFO-924/1506 for the up- and downstream allelic exchange cassettes of C. species CAG138. Respective insert and cassettes were combined to a single fragment by SOEing PCR, digested with SacI and BamHI and ligated into similarly digested, gel purified pJAK112, yielding pFF-320, pFF-314, pFF-321, and pFF-377.
In-frame deletion of the CDIF630_01583-dccA operon (pFF-451).
For in-frame deletion of the CDIF630_01583-dccA operon, homology cassettes were generated using FFO-1733/1741 and FFO-1740/1742. The cassettes were joined by SOEing PCR using FFO-1733/1740, digested with KpnI and SacI to generate sticky ends, and cloned into similarly digested, gel-purified pJAK112.
Cyclic di-GMP reporter (pFF-458/441/442).
As a first step to generate a c-di-GMP responsive reporter plasmid, the riboswitch region from the pilA1 5′-UTR was amplified from C. difficile 630 genomic DNA using FFO-1706/1707 introducing an XbaI restriction site to the 5′-end and an SacI restriction site at the 3′-end. The fragment and pFF-380, a plasmid with a pDSW1728 backbone, containing an fdxA-promoter followed by an XbaI restriction site, located upstream of mCherry and a SacI restriction site downstream of the mCherry start codon, were digested with SacI and XbaI and subsequently ligated, creating a reporter system lacking a start codon for mCherry, pFF-458 (no fluorescence control, pEV).
In a second step, mCherry was PCR-amplified from the same construct with FFO-1710/861, appending a Shine–Dalgarno sequence and a start codon to mCherry and moving the SacI restriction site upstream of the translation start site. pFF-458 and the PCR fragment were digested with SacI and BamHI, gel purified and subsequently ligated, generating pFF-441 (c-di-GMP sensitive reporter, pRS).
To generate an inactive riboswitch control, the pilA1 5′ UTR was amplified from C. difficile 630 gDNA in 2 fragments using FFO-1706/1708 and FFO-1709/1707 introducing an A to G mutation that makes the Riboswitch insensitive to c-di-GMP [44,45]. The fragments were unified by SOEing PCR and both fragment and parent vector pFF-441 were digested with SacI and XbaI (Thermo Scientific) and subsequently ligated resulting in pFF-442 (inactivated c-di-GMP reporter, pRSA70G).
Chromosomal manipulation of C. difficile 630
For chromosomal manipulation, the two-step allelic exchange method was applied as described previously [21,57].
After plasmid conjugation, C. difficile 630 was restreaked at least twice and subsequently screened for single crossover events. Integrations into the modT locus (pFF-173/314/320/321/377/378) were identified by PCR using FFO-360/498, FFO-361/499 and FFO-498/499 and FFO-360/613. Integrations into the pyrE locus (pFF-221/222) were identified using FFO-361/612 and FFO-612/613. Single crossover events for generating Δ01583-dccA (pFF-451) were identified using FFO-1746/361 and FFO-1743/360.
To allow the second recombination event, single crossover integrants were incubated on BHI-agar for approximately 72 h, harvested in 900 μl PBS, and plated in serial dilutions onto CDMM agar plates containing 50 μg/ml 5′-Fluorcytosine. Emerging colonies were restreaked onto BHI and BHI containing thiamphenicol. Cultures growing on BHI, but not on selective BHI were screened for second recombination events.
Successful manipulation at the modT locus was determined by colony PCR using FFO-498/499 and integrity of homology and the knockout region was verified by Sanger sequencing (FFO-498, 499, 482, 483). Similarly, double crossover recombinants at the pyrE locus were identified using FFO-612/613 and verified by Sanger sequencing (FFO-610, 611, 612, and 613). For sequencing of the 01583-dccA operon knockout, FFO-1743, 1746, and FFO-1749 were used.
Hot phenol extraction of total RNA and DNase I digestion
For collection, samples were mixed with ice-cold Stop Mix (95% EtOH, 5% acidic phenol) equivalent to 1/5 of the culture volume and subsequently frozen in liquid N2. For RNA extraction, samples were thawed on ice, centrifuged for 20 min at 4,000 × g, 4°C. Pellets were suspended in 600 μl of 10 mg/ml lysozyme in TE buffer (pH 8) and digested for 10 min at 37°C, and 60 μl of 10% (w/V) SDS was added, samples were mixed by inversion and incubated at 64°C for 1 to 2 min. Subsequently, 66 μl of 3 M NaOAc, pH 5.2 with 1 mM EDTA were added and mixed by inversion, followed by addition of 750 μl of acidic phenol for RNA extraction (Roth). Samples were mixed by shaking and incubated at 64°C in a water bath for 6 min with intermediate mixing. Phases were separated by centrifuging at 4°C, 16,000 × g for 15 min and the aqueous phase was transferred to a Phase Lock Gel tube (Eppendorf), where an equal volume of chloroform was added. Phases were thoroughly mixed and subsequently separated by centrifugation at 16,000 × g, 15°C for 12 min. Again, the aqueous phase was transferred to a new tube and 2 volumes of a 30:1 Ethanol to 3 M NaOAc (pH 6.5) mix was added and mixed by inversion. RNA was precipitated overnight, centrifuged at 16,000 × g, 4°C for 30 min and washed twice with 500 μl ice cold 75% Ethanol, with 10-min centrifugation steps in between. After complete removal of liquid, the pellet was air dried at RT for approximately 5 min and eluted in 50 μl of RNase free water by shaking at 1,000 RPM, 65°C, for 10 min. RNA concentration and purity was assessed with a Nanodrop 2000 spectrophotometer (Thermo Fisher).
Approximately 5 μg of RNA were digested with 5 U DNase I and 10 U RNase Inhibitor (Takara) at 37°C for 1 h. For RNA purification, the sample was adjusted to 100 μl with DEPC-treated ddH2O and transferred to a Phase Lock Gel tube, where it was mixed with 100 μl P:C:I for RNA (Roth) by inversion. Phases were separated by centrifugation at 16,000 × g, 4°C for 15 min. The aqueous phase was transferred to a new tube and precipitated overnight after adding 2.5 volumes of 30:1 Ethanol to NaOAc (pH 6.5) and mixing. Precipitated RNA was centrifuged for 30 min at 16,000 × g, 4°C, washed twice with 200 μl 75% EtOH with 10-min centrifugation steps at 16,000 × g, 4°C, dried for 5 min after complete liquid removal and eluted in 20 μl DEPC-treated water by shaking for 5 min at 1,000 RPM, 65°C.
Rifampicin assay
To inhibit transcription of growing cultures, Rifampicin was added to C. difficile WT cells grown in TYG medium to mid-exponential phase, to a final concentration of 200 μg/ml per culture and mixed by swirling. RNA samples were aliquoted at the indicated time points and samples were fixed by adding ice-cold stop mix, inversion and immediate freezing in liquid nitrogen. RNA was extracted as indicated in the hot phenol protocol.
Northern blotting
For northern blot analysis, 3 to 6 μg of RNA were suspended in 2× Gel loading buffer II (95% formamide, 18 mM EDTA, 0.025% SDS, 2% bromophenol blue), boiled for 5 min at 95°C and subsequently cooled for 5 min on ice. After separation on a 6% polyacrylamide (PAA) gel containing 7 M Urea for 2 h at 300 V, RNA was transferred to a Hybond N+ membrane (GE Healthcare) in 1× TBE using a wet blotting chamber for 1 h at 50 V, 4°C and subsequently cross-linked at 254 nm (0.12 J/cm2).
Membranes were pre-hybridized in Roti Hybri-Quick buffer (Roth) for 1 h at 42°C. RNA of interest was detected with [γ-32P]-labeled oligonucleotides by overnight hybridization at 42°C. A list of all oligonucleotides used in this study can be found in supplementary S4 Dataset. The membrane was washed sequentially in 5×, 1×, and 0.5× SSC containing 0.1% SDS, dried and finally exposed on a phosphor screen for 1 to 7 days. Signals were visualized using a Typhoon FLA7000 phosphor imager. Band intensities were assessed using ImageJ v1.52u [58].
Blots were stripped with 0.1% SDS in distilled, boiled water for 15 min and subsequently washed in distilled water. Blots were hybridized in Roti Hybri-Quick buffer before adding a new oligonucleotide probe.
Oligonucleotide labeling
For [γ-32P] 5′-labeling, 10 pmol of DNA-oligonucleotide probes were incubated with 5 U T4 Polynucleotide Kinase (NEB) together with 1 μl [γ-32P]-ATP (10 μCi/μl) in a volume of 10 μl. After 1 h incubation at 37°C, 10 μl of sterile, DNase- and RNase free water was added and radiolabeled oligonucleotides were separated from free [γ-32P]-ATP using a sephadex G-25 column (Cytiva) according to the manufacturer’s instructions.
In vitro transcription of RNA, RNA labeling, and in-line probing (ILP)
ILP assays were performed as described previously [53]. The short and the long isoform of ModT were PCR amplified using FFO-1235/1089 and FFO-1235/1088 to add a T7 promoter. RNA was in vitro transcribed using the T7 Megascript RT Kit (Thermo Scientific), resolved on a 6% PAA, 7 M Urea gel, excised and eluted overnight in 750 μl RNA elution buffer (0.1 M NaOAc, 0.1% SDS, 10 mM EDTA) at 1,000 RPM, 8°C. RNA was purified by adding 750 μl P:C:I for RNA (Roth), thoroughly mixed and phases were separated by centrifugation. The aqueous phase was transferred to a new tube and RNA was precipitated by adding 1 ml 30:1 EtOH:NaOAc (pH 6.5).
Approximately 50 pmol of in vitro transcribed RNAs were dephosphorylated by CIP (NEB) treatment, purified and precipitated as described before and 20 pmol of eluted RNAs were radioactively 5′-labeled using 10 U T4 Polynucleotide kinase (Thermo Scientific) and 2 μl [γ-32P]-ATP (10 μCi/μl) in 1× PNK Buffer A (Thermo Scientific) in a total volume of 20 μl for 30 min at 37°C. Labeled RNA was purified using a sephadex G-50 column (Cytiva) and a denaturing PAA gel, isolated by overnight elution from the gel and purified by organic phase separation and precipitation of the aqueous phase as described above.
For ILP reactions, labeled ModT was denatured at 95°C for 1 min and subsequently chilled on ice for 5 min, and 0.04 pmol/μl ModT was incubated in a buffer containing final concentration of 100 mM KCl, 20 mM MgCl2, 50 mM Tris-HCl for approximately 40 h at RT in a volume of 15 μl. Control reactions were set up in RNase free water. For a T1 ladder, 0.01 U/μl of RNaseT1 (Thermo Scientific) was incubated with 0.02 pmol/μl of labeled ModT RNA for 5 min at 37°C in a volume of 15 μl. For the alkaline hydrolysis control reaction, 0.02 pmol/μl ModT was heated to 95°C for 5 min in sodium bicarbonate (alkaline hydrolysis) buffer, pH 9 (Thermo Scientific). All reactions were stopped by adding an equal volume of 2× colorless loading dye (10 M Urea, 1.5 mM EDTA). Finally, 10 μl of the quenched reactions was separated on 60-cm long denaturing PAA gels at 40 W, which were dried onto whatman paper for 2 h at 80°C. The radioactive signal was exposed to a phosphor screen, which was read out with a Typhoon FLA 7000 imager (Fuji series).
DMS-structure probing and sequencing
In vitro DMS-structure probing.
For in vitro dimethyl sulfate structure probing (DMS-structure probing), a PCR template was amplified from C. difficile 630 gDNA using a high-fidelity Fusion DNA-polymerase (Mobidiag) with the primers od1192 and od1193 for the long ModT isoforms, and od1192 and od1194 for the short ModT isoform, appending a T7 promoter sequence to the template.
PCR templates were used for in vitro transcription reactions as described above, which were subsequently purified over a silica column and eluted in RNase free water. RNA size and integrity was verified on a 7 M Urea 4% PAA gel.
The RNA stock was denatured for 2 min at 95°C and subsequently chilled on ice for 5 min, and 50 ng of each RNA transcript was allowed to fold for 15 min at 37°C in a “physiological buffer” (20 mM HEPES-NaOH (pH 7.5), 140 mM KCl, 2 mM MgCl2). DMS diluted in EtOH was added to the folded RNAs (final concentration 85 mM) and incubated at 37°C for 6 min. For each condition, an EtOH-only control was included. Reactions were quenched on ice by adding 200 mM final of β-mercaptoethanol (Sigma-Aldrich) and 3 volumes of TRI reagent LS (Sigma Aldrich).
RNA was extracted according to the manufacturer’s instuctions. Briefly, 0.2 volumes of chloroform was added, followed by thorough mixing. Samples were centrifuged at 12,000 × g for 15 min, 4°C and the aqueous phase was transferred to a new tube in which the RNA was precipitated by adding 1 volume of isopropanol and 1 μl of Glycoblue (Invitrogen) followed by a 30 min incubation at −80°C. RNA was centrifuged at 12,000 × g, 4°C for 30 min and the pellets were washed twice with 1 ml of 75% EtOH, air dried, and eluted in 15 μl water.
For cDNA synthesis, half the volume of RNA (7.5 μl) was heated to 65°C together with 1.5 μl 100 μm primer (LongModT_Rv for the long ModT isoform and ShortModT_Rv for short ModT isoform) for 5 min and used for reverse transcription with the Marathon-RT [59] in RT-buffer (50 mM Tris-HCl (pH 8.3), 200 mM KCl, 5 mM DTT, 20% glycerol, 1 mM MnCl2, 0.5 mM dNTP, and 8 U RNaseIn) for 8 h at 42°C.
cDNAs were diluted to 1/8 with nuclease-free water, and 5 μl the diluted cDNA was used as template in 50 μl PCR reaction using the Q5 High-Fidelity DNA Polymerase (NEB) and the primers od1195 and od1194 for the long ModT isoform, and od1195 and od1197 for the short ModT isoform, appending Nextera sequencing adapter sequences HF and HR to the amplicons extremities. Cycling conditions were 2 min at 98°C then 25 cycles of 10 s at 98°C, 15 s at 55°C, and 15 s at 72°C then 72°C for 5 min, and 5 μl of PCR products were verified on a 1.5% agarose gel and the rest was column purified using NucleoSpin Gel and PCR Clean-up kits, Macherey-Nagel according to the manufacturer’s instructions.
A final indexing PCR was carried out using Illumina Nextera DNA CD indexes (96 Indexes, 96 Samples, Illumina). Reaction conditions were 80 ng of purified PCR product, 1 × Q5 reaction buffer, 0.2 mM dNTPs, 2.5 μl of indexing primer, 0.02 U/μl of Q5 polymerase in a final volume of 14 μl. Indexed PCR products were verified on a 1.5% agarose gel, pooled together in an equimolar ratio, before final purification on a 1.5% agarose gel. The pooled indexed sequencing library was quantified using the NEBNext library Quant Kit for Illumina and paired-end PE150 sequencing was carried out on an Illumina Novaseq instrument (Novogene). DMS-MaP data was trimmed using cutadapt v 1.18 [59] and aligned to the reference sequence using bowtie2 [60]. cutadapt parameters were “–nextseq-trim 25 –maxn 0 -g gtgatatttaggtctgtg -G gatatttagctgacctgg.” bowtie2 parameters were “-D 20 -R 3 -N 1 -L 15 -i S,1,0.50.” Further analysis was carried out using the rf-count and rfnorm modules of RNA Framework package v2.7.2 [61]. rfcount parameters were “—count-mutations–only-mut G>Y;A>B;C>D;T>V -q 30” and a sample specific mask file was included to ignore counting at primer binding sites. rf-norm parameters were “-rb AC -sm 3 -nm 2 –remap-reactivities,” meaning that DMS reactivities were calculated by subtracting background mutations in the untreated sample and normalized using 90% Winsorizing.
The match between predicted RNA structures and DMS reactivities was carried out using python v 3.12.3 with the sklearn.metrics.roc_auc_score function from the scikit-learn v 1.5.1 package.
In vivo DMS-structure probing.
C. difficile 630 WT cultures were inoculated in a time-matched manner, so that the structure probing procedure was carried out at the same time for all cultures. For native DMS-probing analysis, 3 ml (~2 to 5 ODs) of culture were treated with 150 μl of 3.4 M DMS in EtOH (final concentration 170 mM) or an equal volume of EtOH for 6 min at 37°C. The reaction was quenched by adding 150 μl of β-mercaptoethanol 14 M (Sigma-Aldrich) and transferring cells on ice. Cells were centrifuged at 5,000 × g for 10 min, 4°C and the pellet was dissolved in 1 ml of TRI reagent (Sigma Aldrich). To support lysis, the ice-cold samples were transferred into tubes containing 500 mg of glass beads and subjected to 3 grinding cycles (30 s, 6 m/s) in a FastPrep (MP Biomedicals) beadbeater with cooling on ice for 2 min between each cycle. Samples were centrifuged for 10 min at 10,000 × g, 4°C. The bead-free supernatant was transferred to a new tube and RNA was extracted according to the manufacturer’s instructions, and eluted in 15 μl RNase free water.
For deproteinated lysate DMS-probing analysis, 6 ml of culture (~2 to 5 ODs) was centrifuged at 5,000 × g for 15 min, 4°C and the bacterial pellet was dissolved in 1 ml of lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM PMSF, 0.2% Triton ×100, 200 U/ml RNaseIn). Cells were lysed mechanically at 4°C, and the lysate was clarified by centrifugation 5 min at 10.000 × g, 4°C; 110 μl of SDS 10% and 50 μl of proteinase K (NEB) were added to 1 ml of clear lysate and the mixture was incubated at 37°C for 30 min with gentle shaking. The digested lysate was equally distributed in 2 microtubes, one added with 50 μl of DMS (85 mM in EtOH) and one added with 50 μl of EtOH, and the incubation was prolonged at 37°C for 6 min. The reaction was quenched by adding 50 μl of β-mercaptoethanol 14 M and transfer on ice, and 1.5 ml of TRI reagent LS was added and the RNA was extracted according to the manufacturer’s instructions, and eluted in 15 μl RNase free water.
Approximately 8 μg of each probed RNA sample (native or deproteinated) were treated with 2 μl Turbo DNase (Thermo Scientific) with 5 μl 10× Turbo DNase buffer and 12 U RNaseIn in a total volume of 50 μl for 30 min at 37°C and subsequently purified over a silica column with NTC buffer (Macherey-Nagel); 1 μg DNase-digested total RNA was used for cDNA synthesis using the Marathon-RT with the conditions indicated above (using the primer LongModT_Rv for the long ModT isoform specifically and ShortModT_Rv for the short ModT isoform, which includes ModT long). cDNAs were purified over a silica column with NTC buffer (Macherey-Nagel). Half of the purified cDNA was PCR-amplified with the Q5 High-Fidelity DNA Polymerase using HF_ModT_Fw and HR_ShortModT_Rv as previously described. PCR products were analyzed on a 1.5% agarose gel and purified on silica column (Macherey-Nagel). Indexing PCR, library preparation, and data analysis was carried out as described in the previous section.
Reverse transcription and quantitative PCR
For cDNA strand synthesis, 1 μg of DNase I digested RNA was reverse transcribed using the M-MLV reverse transcriptase Kit (Invitrogen) with random hexamer primers (Invitrogen) according to the manufacturer’s instructions.
qPCR reactions were performed using 1 μl of 20-fold diluted cDNA and with 2x Takyon NO ROX SYBR Master Mix blue TTP (Eurogentec) with 20 nM primers in a total volume of 10 μl. Each reaction was carried out at least in technical duplicates per biological replicate. Fold change was calculated with the 2-ΔΔCT method [62] using 16S rRNA as a housekeeping reference.
RNA sequencing
Samples for RNA sequencing were collected from C. difficile WT and ΔmodT cultures grown in TYG medium to EP (3, 5h) and to TP (9 h). RNA was extracted using the hot phenol method, DNase I digested and purified as indicated above. cDNA library preparation and sequencing were performed by Vertis Biotechnologie AG. RNA samples were examined by capillary electrophoresis. Ribosomal RNA was depleted using an in-house developed protocol. The ribodepleted RNA samples were fragmented using ultrasound (1 pulse of 30 s at 4°C) and an oligonucleotide adapter was ligated to the 3′ end of the RNA molecules. First-strand cDNA synthesis was performed using M-MLV reverse transcriptase and the 3′ adapter as primer. The first-strand cDNA was purified and the 5′ Illumina TruSeq sequencing adapter was ligated to the 3′ end of the antisense cDNA and the resulting cDNA was PCR-amplified (15 cycles) to about 10 to 20 ng/μl using a high fidelity DNA polymerase. The cDNA was purified using the Agencourt AMPure XP kit (Beckman Coulter Genomics) and analyzed by capillary electrophoresis to estimate library quality and concentration. For Illumina NextSeq sequencing, the samples were pooled in approximately equimolar amounts. The cDNA pool was size fractionated in the size range of 200 to 600 bp using a preparative agarose gel. An aliquot of the size fractionated pool was analyzed by capillary electrophoresis for quality control. The cDNA pool was single-read sequenced on an Illumina NextSeq 500 system using 75 bp read length.
Bioinformatic analysis of RNA sequencing
Raw reads of RNA-seq libraries were trimmed and filtered using BBDuk (ktrim = r, k = 23, mink = 11, hdist = 1, tpe, tbo, qtrim = r, trimq = 20, minlength = 25). Mapping and read counting was conducted with READemption 0.4.3 (READemption align:—poly_a_clipping,—realign, READemption gene_quanti:—skip_antisense,—no_count_splitting_by_gene_no) [63]. Reads were mapped to C. difficile 630 CP010905.2 with additional annotations of UTRs and ncRNAs. Differential expression analysis was performed with edgeR [64]. Normalization factors were calculated using the trimmed means of M values method (TMM). All features with less than 10 reads in at least 2 libraries were excluded from the analysis.
Fluorescence quantification by flow cytometry
At the respective time point, 200 μl of each sample was transferred to a 96-well plate and centrifuged at 4,500 × g, 4°C for 10 min. Supernatant was removed using a multichannel pipette and bacterial pellets were suspended in 150 μl of 4% PFA and fixed at RT in a dark plate for 30 min. Bacteria were washed 3 times in 200 μl PBS with centrifuging steps as indicated above. The final cell pellet was suspended in 30 μl PBS containing 1 μg/ml DAPI and kept overnight in the dark at 4°C to allow full fluorophore maturation. The next day, samples were centrifuged, supernatant was removed and pellets were thoroughly suspended in 200 μl of PBS. Cells were diluted to an appropriate concentration and red fluorescence (λex = 561 nm, signal detection with the custom “PE-Texas Red” bandwidth filter detecting 615 +/−20 nm) was determined using a Novocyte Quanteon flow cytometer (Agilent) from 100,000 events, selecting exclusively DAPI-positive cells (λex = 405 nm, applying the “Pacific-Blue” filter (detecting 445 +/−45 nm) and a FSC-H (>5,000 AU) cutoff to discriminate cells from false positive debris events. The mean fluorescence of the selected events was taken as readout. To normalize fluorescence, the mean fluorescence from the inactivated riboswitch (pRSA70G) was subtracted from the functional, c-di-GMP sensitive reporter (pRS) in each respective background. Fluorescence of all strains carrying the nonfluorescent control (pEV) was measured, but was not included in the calculation.
Supporting information
S3 Dataset. SigB vs. RNAseq: Differentially expressed genes (DEGs) in the modT mutant that were also up-regulated after sigB induction (log2FC bigger or equal to 1) [35].
Sporulation vs. RNAseq: Differentially expressed genes (DEGs) in the modT mutant that are part of the sporulation pathway. # Indicates genes that are induced during sporulation by Spo0A, σF, σE, σG, or σK [39].
https://doi.org/10.1371/journal.pbio.3002948.s003
(XLSX)
S1 Fig. Analysis of ModT RNA features.
(A) Read profiles obtained from differential RNA-seq (dRNA-seq) and RNAtag-seq libraries. For dRNA-seq, half of the libraries are treated with terminator exonuclease (TEX+) which degrades processed (non-triphosphorylated) 5′ ends. This differential treatment results in a relative read enrichment for primary transcripts (5′-PPP) in the TEX+ libraries, whereas it results in a relative read enrichment for processed transcripts in the TEX- libraries. As a result, it allows the identification of transcript start sites (TSS) and 5′ processing sites. For RNAtag-seq, adapters for sequencing are ligated to RNA 3′ ends, which leads to enriched read coverage at 3′ ends of transcripts, thus facilitating the annotation of transcription termination sites (TTS). The genome coordnates correspond to genome annotation CP010905.2. The data underlying this figure can be found in https://doi.org/10.1073/pnas.2103579118 and under the accession number GSE155167. (B) ModT expression profile in C. difficile 630 across different growth stages. Total RNA was extracted from C. difficile 630 grown in TY medium supplemented with indicated carbon sources at indicated time points (same RNA as in Fig 1C). ModT transcript was detected by northern blot using a radioactively labeled DNA probe (FFO-358) specific for the extended 3′ end of the long ModT isoform (ModT(L)). 5S rRNA served as loading control. (C) Quantification of RNA half-lives based on 3 biological replicates, as displayed in Fig 1D. The calculated half-life of each transcript is shown above. The underlying data can be found in S5 Dataset. (D) GC content of noncoding RNA elements. Analysis was based on a manually curated genome annotation (CP010905.2) for C. difficile 630 that includes annotation of noncoding gene loci [26]. The dotted line indicates the average GC content of the genome. The underlying data can be found in S5 Dataset.
https://doi.org/10.1371/journal.pbio.3002948.s007
(TIF)
S2 Fig. In vitro and in vivo DMS structure probing of ModT isoforms.
(A–D) In-line probing reactions (ILP) of in vitro transcribed, 5′ 32P-labeled ModT(S) and ModT(L) isoforms, resolved on denaturing (7 M Urea) 10% PAA gels (A, B). In parallel, the same samples were resolved on a 6% PAA-7M Urea gel (C, D) to increase resolution of residues in the 3′ end of both isoforms. RNA was left untreated as control (Ctr). Denatured RNA, digested by RNase T1 (T1) or cleaved by alkaline hydrolysis (OH), served as ladder. Base-paired regions are indicated according to the secondary structure model shown in Fig 2A. (E) Heatmap displaying correlations of all DMS reactivity samples calculated using the Pearson’s correlation coefficient and hierarchical clustering using the single linkage algorithm. (F) The Δreactivity values for each nucleotide position obtained for the indicated conditions were plotted onto the C. difficile ModT consensus secondary structure model shown in Fig 2A–2C. For “in vivo Native → Δ(3h-9h),” the profiles of ModT(L) and ModT(S) were combined to obtain the mean reactivity and standard deviation at these 2 time points. For “ModT(L) → Δ(Deproteinated–Native),” the mean subtracted reactivity and its standard deviation was calculated from all growth stages (3 h, 9 h, and 24 h). (G) Heatmap displaying ROC-AUC scores demonstrating the fit of DMS reactivities from each sample to every structure model depicted in Fig 2A–2C. An ROC-AUC score of 0.5 signifies a random association of reactivity and base pairing status, and 1 indicates the perfect agreement.
https://doi.org/10.1371/journal.pbio.3002948.s008
(TIF)
S3 Fig. ModT regulates bacterial transition into the stationary phase.
(A) qPCR quantification of expression levels of ModT as well as immediate up- and downstream located genes in wild-type and ΔmodT during exponential phase (EP, 3, 5 h) and transition phase (TP, 9 h). Unpaired t test with Welch correction. The underlying data can be found in S5 Dataset. (B) Northern blot-based expression analysis of ModT in WT, ΔmodT and complemented strains across different growth stages in TYG medium. 5S rRNA served as loading control. Depicted is a representative of 3 biological replicates. (C) Maximum optical density (OD) of wild type and ΔmodT grown in TY medium supplemented with indicated carbon sources (n = 3 biological replicates). Significance was determined with an unpaired t test with Welch correction. The underlying data can be found in S5 Dataset. (D) Growth curves of C. difficile wild type and ΔmodT indicating the time points of RNA isolation for sequencing. EP = exponential phase, 3, 5 h; TP = transition phase, 9 h. The underlying data can be found in S5 Dataset. (E) Venn diagram displaying overlap of up- and down-regulated genes in ΔmodT relative to wild type between exponential phase (EP, 3, 5 h) and transition phase (TP, 9 h). (F) Venn diagram displaying overlap of the SigB regulon [36] (log2FC≥1, p ≤ 0.05) and differentially expressed genes in ΔmodT during exponential phase (EP, 3, 5 h) and transition phase (TP, 9 h) (log2FC ≥1/≤-1, FDR ΔmodT are depicted below. (G) Northern blot-based expression analysis of ModT, tRNA His, tRNA Ala, and 5S RNA in wild type and ΔmodT across different growth stages in TYG medium. Depicted is a representative of 3 biological replicates.
https://doi.org/10.1371/journal.pbio.3002948.s009
(TIF)
S4 Fig. CFU-based quantification of sporulation frequencies.
(A) and (B) Sporulation frequency in 70:30 medium determined by plate-based assay. Total CFU of spores germinated on BHIS-agar supplemented with 0.1% Taurocholate (top) and of vegetative bacterial cells recovered on BHIS agar (bottom) are displayed in (A). Calculated sporulation frequency based on CFU values (A) is displayed in (B). Significance for data shown in (A) and (B) was determined by two-way ANOVA with Tukey’s multiple comparisons test. Only significant differences with P P P P S5 Dataset. (C) Northern blot-based expression analysis of ModT in WT, ΔmodT and complemented strains across different growth stages in 70:30 sporulation medium. 5S rRNA serves as loading control. Depicted is a representative of 3 biological replicates.
https://doi.org/10.1371/journal.pbio.3002948.s010
(TIF)
S5 Fig. ModT modulates cellular c-di-GMP levels.
(A) Schematic representation of riboswitch reporter. Green box represents the constitutive fdxA (CDIF630_00294)-promoter, the double stem loop with black pin represents the repressing c-di-GMP II riboswitch conformation at low c-di-GMP levels, while the dashed, white pin indicates the open conformation at high c-di-GMP levels, allowing transcription of the downstream mCherry fluorophore. (B) Raw data used for c-di-GMP quantification in Fig 4A. To account for potential variation in promoter activity in the reporter (pRS), the signal from the construct with inactivated Riboswitch (pRSA>G) was subtracted from the actual reporter pRS. In addition, an empty vector control (pEV) was used to normalize for potential differences in baseline fluorescence due to different mutant backgrounds. n = 3 biological replicates per strain. Significance was determined by two-way ANOVA with Tukey’s multiple comparisons test. Only significant differences with P P P S5 Dataset.
https://doi.org/10.1371/journal.pbio.3002948.s011
(TIF)
S6 Fig. Orthologs from distant phylogenetic families can replace ModT function in C. difficile.
(A) Sequence alignment of ModT orthologs from C. difficile (Cd), P. sordellii (Ps), C. perfringens (Cp), Acetohalobium arabaticum (Aa), and Clostridium sp. CAG:138 (CAG). Color-coding of regions P1-P8 is identical to Fig 2A. (B) Schematic representation of chromosomal modT deletion and replacement of the short ModT isoform in C. difficile 630 with orthologous sequences from P. sordellii (Ps), C. perfringens (Cp), Acetohalobium arabaticum (Aa), and Clostridium sp. CAG:138 (CAG). (C) Northern blot-based expression analysis of ModT in WT, ΔmodT and in C. difficile 630 with ModT orthologue replacements across different growth stages in 70:30 medium. 5S rRNA serves as loading control. Depicted is a representative of 3 biological replicates. (D) Representative phase-contrast micrographs of cultures used for calculating sporulation frequencies in Fig 6C. Scale bar = 5 μm. (E) Plate-based sporulation assay in 70:30 medium for WT, ΔmodT and C. difficile 630 with ModT orthologue replacements. Total CFU of vegetative bacterial cells growing on BHIS agar and total CFU of germinated spores determined in BHIS-agar supplemented with 0.1% Taurocholate. Time points are indicated relative to the onset of the transition phase. Experiments were performed in (n = 3) biological replicates. Significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test. Only significant differences with P P P P S5 Dataset. (F) Calculated sporulation frequency based on CFU and spore values shown in (E). Significance was determined using two-way ANOVA with Dunnett’s multiple comparisons test. Only significant differences with P P P P S5 Dataset. (G) Alignment of promoter region upstream of the transcription start site of modT in C. difficile (Cd), P. sordellii (Ps), C. perfringens (Cp), and C. acetobutylicum (Ca).
https://doi.org/10.1371/journal.pbio.3002948.s012
(TIF)
Acknowledgments
We greatly appreciate the critical and constructive feedback on the manuscript that was provided by Anke Sparmann, Manuela Fuchs, Sara Giddins, and Janet Wackenreuther.
References
- 1.
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157(1):77–94. pmid:24679528 - 2.
Eichner H, Karlsson J, Loh E. The emerging role of bacterial regulatory RNAs in disease. Trends Microbiol. 2022;30(10):959–972. pmid:35379550 - 3.
Nelson JW, Breaker RR. The lost language of the RNA World. Sci Signal. 2017;10(483). pmid:28611182 - 4.
Westermann AJ. Regulatory RNAs in Virulence and Host-Microbe Interactions. Microbiol Spectr. 2018;6(4). pmid:30003867 - 5.
Serganov A, Patel DJ. Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat Rev Genet. 2007;8(10):776–790. pmid:17846637 - 6.
Faure G, Makarova KS, Koonin EV. CRISPR-Cas: Complex Functional Networks and Multiple Roles beyond Adaptive Immunity. J Mol Biol. 2019;431(1):3–20. pmid:30193985 - 7.
Hör J, Matera G, Vogel J, Gottesman S, Storz G. Trans-Acting Small RNAs and Their Effects on Gene Expression in Escherichia coli and Salmonella enterica. EcoSal Plus. 2020;9(1). pmid:32213244 - 8.
Jose BR, Gardner PP, Barquist L. Transcriptional noise and exaptation as sources for bacterial sRNAs. Biochem Soc Trans. 2019. pmid:30837318 - 9.
Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35(3 Pt 2):849–857. - 10.
Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat Struct Biol. 2000;7(6):449–455. pmid:10881189 - 11.
Nagai K, Oubridge C, Kuglstatter A, Menichelli E, Isel C, Jovine L. Structure, function and evolution of the signal recognition particle. EMBO J. 2003;22(14):3479–3485. pmid:12853463 - 12.
Walter P, Blobel G. Signal recognition particle contains a 7S RNA essential for protein translocation across the endoplasmic reticulum. Nature. 1982;299(5885):691–698. pmid:6181418 - 13.
Wallace JG, Zhou Z, Breaker RR. OLE RNA protects extremophilic bacteria from alcohol toxicity. Nucleic Acids Res. 2012;40(14):6898–6907. pmid:22561371 - 14.
Walter P, Blobel G. Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc Natl Acad Sci U S A. 1980;77(12):7112–7116. pmid:6938958 - 15.
Weinberg Z, Lunse CE, Corbino KA, Ames TD, Nelson JW, Roth A, et al. Detection of 224 candidate structured RNAs by comparative analysis of specific subsets of intergenic regions. Nucleic Acids Res. 2017;45(18):10811–10823. pmid:28977401 - 16.
Weinberg Z, Perreault J, Meyer MM, Breaker RR. Exceptional structured noncoding RNAs revealed by bacterial metagenome analysis. Nature. 2009;462(7273):656–659. pmid:19956260 - 17.
Harris KA, Zhou Z, Peters ML, Wilkins SG, Breaker RR. A second RNA-binding protein is essential for ethanol tolerance provided by the bacterial OLE ribonucleoprotein complex. Proc Natl Acad Sci U S A. 2018;115(27):E6319–e28. pmid:29915070 - 18.
Lee ER, Baker JL, Weinberg Z, Sudarsan N, Breaker RR. An allosteric self-splicing ribozyme triggered by a bacterial second messenger. Science. 2010;329(5993):845–848. pmid:20705859 - 19.
Breaker RR, Harris KA, Lyon SE, Wencker FDR, Fernando CM. Evidence that OLE RNA is a component of a major stress-responsive ribonucleoprotein particle in extremophilic bacteria. Mol Microbiol. 2023;120(3):324–340. pmid:37469248 - 20.
Puerta-Fernandez E, Barrick JE, Roth A, Breaker RR. Identification of a large noncoding RNA in extremophilic eubacteria. Proc Natl Acad Sci U S A. 2006;103(51):19490–19495. pmid:17164334 - 21.
Fuchs M, Lamm-Schmidt V, Sulzer J, Ponath F, Jenniches L, Kirk JA, et al. An RNA-centric global view of Clostridioides difficile reveals broad activity of Hfq in a clinically important gram-positive bacterium. Proc Natl Acad Sci U S A. 2021;118(25). - 22.
Soares LW, King CG, Fernando CM, Roth A, Breaker RR. Genetic disruption of the bacterial raiA motif noncoding RNA causes defects in sporulation and aggregation. Proc Natl Acad Sci U S A. 2024;121(6):e2318008121. pmid:38306478 - 23.
Setlow P. Spore Resistance Properties Microbiology spectrum. 2014;2:5. - 24.
Setlow P, Christie G. New Thoughts on an Old Topic: Secrets of Bacterial Spore Resistance Slowly Being Revealed. Microbiol Mol Biol Rev. 2023;87(2):e0008022. pmid:36927044 - 25.
Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, et al. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun. 2012;80(8):2704–2711. pmid:22615253 - 26.
Lamm-Schmidt V, Fuchs M, Sulzer J, Gerovac M, Hör J, Dersch P, et al. Grad-seq identifies KhpB as a global RNA-binding protein in Clostridioides difficile that regulates toxin production. Microlife. 2021;2:uqab004. pmid:37223250 - 27.
Cao X, Zhang Y, Ding Y, Wan Y. Identification of RNA structures and their roles in RNA functions. Nat Rev Mol Cell Biol. 2024. pmid:38926530 - 28.
Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, et al. Non-Coding RNA Analysis Using the Rfam Database. Curr Protoc Bioinformatics. 2018;62(1):e51. pmid:29927072 - 29.
Kalvari I, Nawrocki EP, Ontiveros-Palacios N, Argasinska J, Lamkiewicz K, Marz M, et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021;49(D1):D192–d200. pmid:33211869 - 30.
Tomezsko P, Swaminathan H, Rouskin S. DMS-MaPseq for Genome-Wide or Targeted RNA Structure Probing In Vitro and In Vivo. Methods Mol Biol. 2021;2254:219–238. pmid:33326078 - 31.
Bohn P, Gribling-Burrer AS, Ambi UB, Smyth RP. Nano-DMS-MaP allows isoform-specific RNA structure determination. Nat Methods. 2023;20(6):849–859. pmid:37106231 - 32.
Collins J, Robinson C, Danhof H, Knetsch CW, van Leeuwen HC, Lawley TD, et al. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature. 2018;553(7688):291–294. pmid:29310122 - 33.
Engevik MA, Engevik AC, Engevik KA, Auchtung JM, Chang-Graham AL, Ruan W, et al. Mucin-Degrading Microbes Release Monosaccharides That Chemoattract Clostridioides difficile and Facilitate Colonization of the Human Intestinal Mucus Layer. ACS Infect Dis. 2021;7(5):1126–1142. pmid:33176423 - 34.
Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114. pmid:24445449 - 35.
Boekhoud IM, Michel AM, Corver J, Jahn D, Smits WK. Redefining the Clostridioides difficile σ(B) Regulon: σ(B) Activates Genes Involved in Detoxifying Radicals That Can Result from the Exposure to Antimicrobials and Hydrogen Peroxide. mSphere. 2020;5(5). - 36.
Kint N, Janoir C, Monot M, Hoys S, Soutourina O, Dupuy B, et al. The alternative sigma factor sigma(B) plays a crucial role in adaptive strategies of Clostridium difficile during gut infection. Environ Microbiol. 2017;19(5):1933–1958. - 37.
Saujet L, Monot M, Dupuy B, Soutourina O, Martin-Verstraete I. The key sigma factor of transition phase, SigH, controls sporulation, metabolism, and virulence factor expression in Clostridium difficile. J Bacteriol. 2011;193(13):3186–3196. pmid:21572003 - 38.
Prossliner T, Agrawal S, Heidemann DF, Sørensen MA, Svenningsen SL. tRNAs Are Stable After All: Pitfalls in Quantification of tRNA from Starved Escherichia coli Cultures Exposed by Validation of RNA Purification Methods. MBio. 2023;14(1):e0280522. pmid:36598190 - 39.
Fimlaid KA, Bond JP, Schutz KC, Putnam EE, Leung JM, Lawley TD, et al. Global analysis of the sporulation pathway of Clostridium difficile. PLoS Genet. 2013;9(8):e1003660. pmid:23950727 - 40.
Purcell EB, McKee RW, McBride SM, Waters CM, Tamayo R. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J Bacteriol. 2012;194(13):3307–3316. pmid:22522894 - 41.
Edwards AN, Willams CL, Pareek N, McBride SM, Tamayo R. c-di-GMP Inhibits Early Sporulation in Clostridioides difficile. mSphere. 2021;6(6):e0091921. pmid:34878288 - 42.
Girinathan BP, Ou J, Dupuy B, Govind R. Pleiotropic roles of Clostridium difficile sin locus. PLoS Pathog. 2018;14(3):e1006940. pmid:29529083 - 43.
Reyes Ruiz LM, King KA, Agosto-Burgos C, Gamez IS, Gadda NC, Garrett EM, et al. Coordinated modulation of multiple processes through phase variation of a c-di-GMP phosphodiesterase in Clostridioides difficile. PLoS Pathog. 2022;18(7):e1010677. pmid:35789350 - 44.
Bordeleau E, Purcell EB, Lafontaine DA, Fortier LC, Tamayo R, Burrus V. Cyclic di-GMP riboswitch-regulated type IV pili contribute to aggregation of Clostridium difficile. J Bacteriol. 2015;197(5):819–832. pmid:25512308 - 45.
Purcell EB, McKee RW, Bordeleau E, Burrus V, Tamayo R. Regulation of Type IV Pili Contributes to Surface Behaviors of Historical and Epidemic Strains of Clostridium difficile. J Bacteriol. 2016;198(3):565–577. pmid:26598364 - 46.
Bordeleau E, Fortier LC, Malouin F, Burrus V. c-di-GMP turn-over in Clostridium difficile is controlled by a plethora of diguanylate cyclases and phosphodiesterases. PLoS Genet. 2011;7(3):e1002039. pmid:21483756 - 47.
Orrell KE, Melnyk RA. Large Clostridial Toxins: Mechanisms and Roles in Disease. Microbiol Mol Biol Rev. 2021;85(3):e0006421. pmid:34076506 - 48.
Shen A, Edwards AN, Sarker MR, Paredes-Sabja D. Sporulation and Germination in Clostridial Pathogens. Microbiol Spectr. 2019;7(6). pmid:31858953 - 49.
Jenal U, Reinders A, Lori C. Cyclic di-GMP: second messenger extraordinaire. Nat Rev Microbiol. 2017;15(5):271–284. pmid:28163311 - 50.
McKee RW, Mangalea MR, Purcell EB, Borchardt EK, Tamayo R. The second messenger cyclic Di-GMP regulates Clostridium difficile toxin production by controlling expression of sigD. J Bacteriol. 2013;195(22):5174–5185. pmid:24039264 - 51.
McKee RW, Harvest CK, Tamayo R. Cyclic Diguanylate Regulates Virulence Factor Genes via Multiple Riboswitches in Clostridium difficile. mSphere. 2018;3(5). pmid:30355665 - 52.
Dar D, Sorek R. High-resolution RNA 3’-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res. 2018;46(13):6797–6805. pmid:29669055 - 53.
Fuchs M, Lamm-Schmidt V, Lenče T, Sulzer J, Bublitz A, Wackenreuter J, et al. A network of small RNAs regulates sporulation initiation in Clostridioides difficile. EMBO J. 2023;42(12):e112858. pmid:37140366 - 54.
Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428(6980):281–286. pmid:15029187 - 55.
Owens RA, Baumstark T. Structural differences within the loop E motif imply alternative mechanisms of viroid processing. RNA. 2007;13(6):824–834. pmid:17438124 - 56.
Edwards AN, Nawrocki KL, McBride SM. Conserved oligopeptide permeases modulate sporulation initiation in Clostridium difficile. Infect Immun. 2014;82(10):4276–4291. pmid:25069979 - 57.
Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol. 2012;78(13):4683–4690. pmid:22522680 - 58.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. pmid:22930834 - 59.
Zhao C, Liu F, Pyle AM. An ultraprocessive, accurate reverse transcriptase encoded by a metazoan group II intron. RNA. 2018;24(2):183–195. pmid:29109157 - 60.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. pmid:22388286 - 61.
Incarnato D, Morandi E, Simon LM, Oliviero S. RNA Framework: an all-in-one toolkit for the analysis of RNA structures and post-transcriptional modifications. Nucleic Acids Res. 2018;46(16):e97. pmid:29893890 - 62.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. pmid:11846609 - 63.
Förstner KU, Vogel J, Sharma CM. READemption-a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics (Oxford, England). 2014;30(23):3421–3423. pmid:25123900 - 64.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England). 2010;26(1):139–140. pmid:19910308 - 65.
Pishdadian K, Fimlaid KA, Shen A. SpoIIID-mediated regulation of σK function during Clostridium difficile sporulation. Mol Microbiol. 2015;95(2):189–208.
ADVERTISEMENT:
Hai, sobat pengemar slots pernahkah mendengar semboyan “slot gaco”? jika tidak, bersiaplah jatuh hati sama konsep ini. slot demo adalah mesin slot yang selalu memberi win. Ya, mesin-mesin ini bisa dikatakan sebagai andalannya tuk membawa pulang hasil. tapi, gimana sih caranya jumpain slot gaco yang benar? Santai Bro, kita bahas tenang aja di sini
Permainan terpercaya saat sekarang satu-satunya di Indonesia yaitu pasti memberikan return tertinggi
Daftar dengan di :
Informasi mengenai KING SLOT, Segera Daftar Bersama king selot terbaik dan terpercaya no satu di Indonesia. Boleh mendaftar melalui sini king slot serta memberikan hasil kembali yang paling tinggi saat sekarang ini hanyalah KING SLOT atau Raja slot paling gacor, gilak dan gaco saat sekarang di Indonesia melalui program return tinggi di kingselot serta pg king slot
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama kdwapp.com
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama jebswagstore.com
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama demoslotgacor.pro
slot demo gacor
slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun demo slot gacor
akun demo slot gacor permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun slot demo gacor
akun slot demo gacor permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun demo slot pragmatic
akun demo slot pragmatic permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun slot demo pragmatic
akun slot demo pragmatic permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun slot demo
akun slot demo permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
akun demo slot
akun demo slot permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
situs slot terbaru
situs slot terbaru permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
slot terbaru
slot terbaru permainan paling top dan garansi imbal balik hasil besar bersama situsslotterbaru.net
suara88 permainan paling top dan garansi imbal balik hasil besar bersama suara88.biz
sumo7777 permainan paling top dan garansi imbal balik hasil besar bersama sumo7777.com
supermoney888 permainan paling top dan garansi imbal balik hasil besar bersama supermoney888.biz
teratai88 permainan paling top dan garansi imbal balik hasil besar bersama teratai88.biz
thor88 permainan paling top dan garansi imbal balik hasil besar bersama thor88.biz
togelhk88 permainan paling top dan garansi imbal balik hasil besar bersama togelhk88.net
topjitu88 permainan paling top dan garansi imbal balik hasil besar bersama topjitu88.net
totosloto88 permainan paling top dan garansi imbal balik hasil besar bersama totosloto88.com
trisula888 permainan paling top dan garansi imbal balik hasil besar bersama trisula888.biz
udangbet88 permainan paling top dan garansi imbal balik hasil besar bersama udangbet88.net
via88 permainan paling top dan garansi imbal balik hasil besar bersama via88.biz
virusjp88 permainan paling top dan garansi imbal balik hasil besar bersama virusjp88.net
warga888 permainan paling top dan garansi imbal balik hasil besar bersama warga888.biz
waw88 permainan paling top dan garansi imbal balik hasil besar bersama waw88.biz
winjitu88 permainan paling top dan garansi imbal balik hasil besar bersama winjitu88.net
wisdom88 permainan paling top dan garansi imbal balik hasil besar bersama wisdom88.biz
wnitogel88 permainan paling top dan garansi imbal balik hasil besar bersama wnitogel88.com
yoyo888 permainan paling top dan garansi imbal balik hasil besar bersama yoyo888.biz
validtoto88 permainan paling top dan garansi imbal balik hasil besar bersama validtoto88.com
sule999 permainan paling top dan garansi imbal balik hasil besar bersama sule999.com
sule88 permainan paling top dan garansi imbal balik hasil besar bersama sule88.org
ss888bet permainan paling top dan garansi imbal balik hasil besar bersama ss888bet.com
sia77 permainan paling top dan garansi imbal balik hasil besar bersama sia77.info
seluang88 permainan paling top dan garansi imbal balik hasil besar bersama seluang88.com
satu88 permainan paling top dan garansi imbal balik hasil besar bersama satu88.biz
satu777 permainan paling top dan garansi imbal balik hasil besar bersama satu777.asia
rp88 permainan paling top dan garansi imbal balik hasil besar bersama rp88.biz
rp88 permainan paling top dan garansi imbal balik hasil besar bersama rp88.asia
rp88 permainan paling top dan garansi imbal balik hasil besar bersama rp77.live
qiuqiu88 permainan paling top dan garansi imbal balik hasil besar bersama qiuqiu88.biz
pt88 permainan paling top dan garansi imbal balik hasil besar bersama pt88.org
pt77 permainan paling top dan garansi imbal balik hasil besar bersama pt77.info
produk88 permainan paling top dan garansi imbal balik hasil besar bersama produk88.asia
mt88 permainan paling top dan garansi imbal balik hasil besar bersama mt88.org
mt77 permainan paling top dan garansi imbal balik hasil besar bersama mt77.biz
menang66 permainan paling top dan garansi imbal balik hasil besar bersama menang66.biz
latobet888 permainan paling top dan garansi imbal balik hasil besar bersama latobet888.org
kedai96 permainan paling top dan garansi imbal balik hasil besar bersama kedai96.org
kedai188 permainan paling top dan garansi imbal balik hasil besar bersama kedai188.biz
ids88 permainan paling top dan garansi imbal balik hasil besar bersama ids88.biz
hp88 permainan paling top dan garansi imbal balik hasil besar bersama hp88.org
hp77 permainan paling top dan garansi imbal balik hasil besar bersama hp77.org
gm88 permainan paling top dan garansi imbal balik hasil besar bersama gm88.asia
gm77 permainan paling top dan garansi imbal balik hasil besar bersama gm77.net
final888 permainan paling top dan garansi imbal balik hasil besar bersama final888.org
duit88 permainan paling top dan garansi imbal balik hasil besar bersama duit88.asia
duit168 permainan paling top dan garansi imbal balik hasil besar bersama duit168.biz
divisi88 permainan paling top dan garansi imbal balik hasil besar bersama divisi88.org
dewi500 permainan paling top dan garansi imbal balik hasil besar bersama dewi500.biz
devil88 permainan paling top dan garansi imbal balik hasil besar bersama devil88.info
cuputoto88 permainan paling top dan garansi imbal balik hasil besar bersama cuputoto88.com
cukongbet88 permainan paling top dan garansi imbal balik hasil besar bersama cukongbet88.asia
bom888 permainan paling top dan garansi imbal balik hasil besar bersama bom888.biz
bintaro888 permainan paling top dan garansi imbal balik hasil besar bersama bintaro888.info
askasino88 permainan paling top dan garansi imbal balik hasil besar bersama askasino88.org
999aset permainan paling top dan garansi imbal balik hasil besar bersama 999aset.com
afb77 permainan paling top dan garansi imbal balik hasil besar bersama afb77.biz
aset99 permainan paling top dan garansi imbal balik hasil besar bersama aset99.biz
bendera77 permainan paling top dan garansi imbal balik hasil besar bersama bendera77.biz
bendera888 permainan paling top dan garansi imbal balik hasil besar bersama bendera888.com
coco88 permainan paling top dan garansi imbal balik hasil besar bersama coco88.org
cuma77 permainan paling top dan garansi imbal balik hasil besar bersama cuma77.biz
cuma88 permainan paling top dan garansi imbal balik hasil besar bersama cuma88.org
dwv88 permainan paling top dan garansi imbal balik hasil besar bersama dwv88.org
fafajp88 permainan paling top dan garansi imbal balik hasil besar bersama fafajp88.com
gemar88 permainan paling top dan garansi imbal balik hasil besar bersama gemar88.biz
gocap88 permainan paling top dan garansi imbal balik hasil besar bersama gocap88.info
gocaptoto permainan paling top dan garansi imbal balik hasil besar bersama gocaptoto.asia
hakabet88 permainan paling top dan garansi imbal balik hasil besar bersama hakabet88.com
hwtoto88 permainan paling top dan garansi imbal balik hasil besar bersama hwtoto88.org
ina77 permainan paling top dan garansi imbal balik hasil besar bersama ina77.biz
ina88 permainan paling top dan garansi imbal balik hasil besar bersama ina88.info
jingga8888 permainan paling top dan garansi imbal balik hasil besar bersama jingga8888.com
juragan777 permainan paling top dan garansi imbal balik hasil besar bersama juragan777.asia
kastil77 permainan paling top dan garansi imbal balik hasil besar bersama kastil77.info
kebo888 permainan paling top dan garansi imbal balik hasil besar bersama kebo888.biz
kkwin77 permainan paling top dan garansi imbal balik hasil besar bersama kkwin77.com
kokoslot88 permainan paling top dan garansi imbal balik hasil besar bersama kokoslot88.asia
luckydf88 permainan paling top dan garansi imbal balik hasil besar bersama luckydf88.org
microstar888 permainan paling top dan garansi imbal balik hasil besar bersama microstar888.biz
monperatoto88 permainan paling top dan garansi imbal balik hasil besar bersama monperatoto88.com
mpo1122 permainan paling top dan garansi imbal balik hasil besar bersama mpo1122.biz
mpo122 permainan paling top dan garansi imbal balik hasil besar bersama mpo122.biz
mpopelangi88 permainan paling top dan garansi imbal balik hasil besar bersama mpopelangi88.com
pamanslot88 permainan paling top dan garansi imbal balik hasil besar bersama pamanslot88.biz
panel88 permainan paling top dan garansi imbal balik hasil besar bersama panel88.org
paragon77 permainan paling top dan garansi imbal balik hasil besar bersama paragon77.biz
paragon888 permainan paling top dan garansi imbal balik hasil besar bersama paragon888.info
pion77 permainan paling top dan garansi imbal balik hasil besar bersama pion77.biz
prada88 permainan paling top dan garansi imbal balik hasil besar bersama prada88.asia
prada888 permainan paling top dan garansi imbal balik hasil besar bersama prada888.com
qqslot88slot permainan paling top dan garansi imbal balik hasil besar bersama qqslot88slot.com
rejekibet88 permainan paling top dan garansi imbal balik hasil besar bersama rejekibet88.com
rezekibet88 permainan paling top dan garansi imbal balik hasil besar bersama rezekibet88.org
sensa77 permainan paling top dan garansi imbal balik hasil besar bersama sensa77.biz
sensa888 permainan paling top dan garansi imbal balik hasil besar bersama sensa888.biz
singajp88 permainan paling top dan garansi imbal balik hasil besar bersama singajp88.com
sr77 permainan paling top dan garansi imbal balik hasil besar bersama sr77.org
sr88 permainan paling top dan garansi imbal balik hasil besar bersama sr88.org
surya77 permainan paling top dan garansi imbal balik hasil besar bersama surya77.biz
surya88 permainan paling top dan garansi imbal balik hasil besar bersama surya88.asia
tajir77 permainan paling top dan garansi imbal balik hasil besar bersama tajir77.info
tajir88 permainan paling top dan garansi imbal balik hasil besar bersama tajir88.biz
toto122 permainan paling top dan garansi imbal balik hasil besar bersama toto122.com
toto123 permainan paling top dan garansi imbal balik hasil besar bersama toto123.biz
uangvip88 permainan paling top dan garansi imbal balik hasil besar bersama uangvip88.com
wajik77 permainan paling top dan garansi imbal balik hasil besar bersama wajik77.asia
777neko permainan paling top dan garansi imbal balik hasil besar bersama 777neko.org
88judi permainan paling top dan garansi imbal balik hasil besar bersama 88judi.net
99judi permainan paling top dan garansi imbal balik hasil besar bersama 99judi.org
abcslot88 permainan paling top dan garansi imbal balik hasil besar bersama abcslot88.asia



